Tema 8 - Farmacocinética Cuantitativa - 1/10-
Clase del 1/10 Presentación
Preguntas a responder:
- ¿A qué velocidad se absorbe un fármaco después de administrarlo?
- ¿Qué concentración alcanzará en plasma y durante cuánto tiempo se mantendrá en un nivel terapéutico?
- ¿Con qué intervalo debemos administrar la medicación para que sea eficaz y segura?
- ¿Cuál es la dosis óptima para una especie determinada?
La farmacocinética cuantitativa nos permite, mediante modelos matemáticos sencillos, conocer los siguientes parámetros farmacocinéticos:
- Velocidad de absorción
- Cantidad de fármaco absorbido
- Modo de distribución del fármaco
- Velocidad de eliminación
- …
Establecer un régimen posológico:
- Dosis óptima
- Intervalo de administración
- Duración del tratamiento
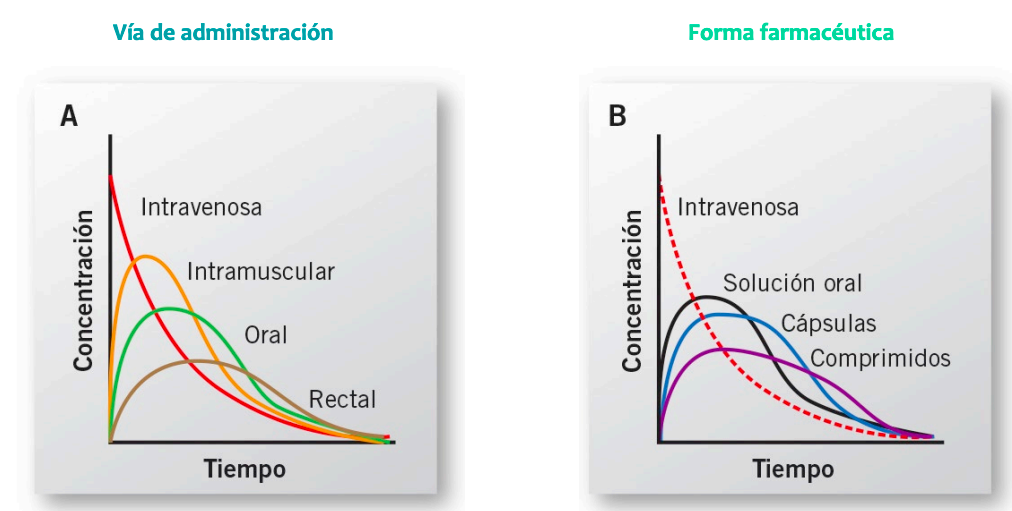
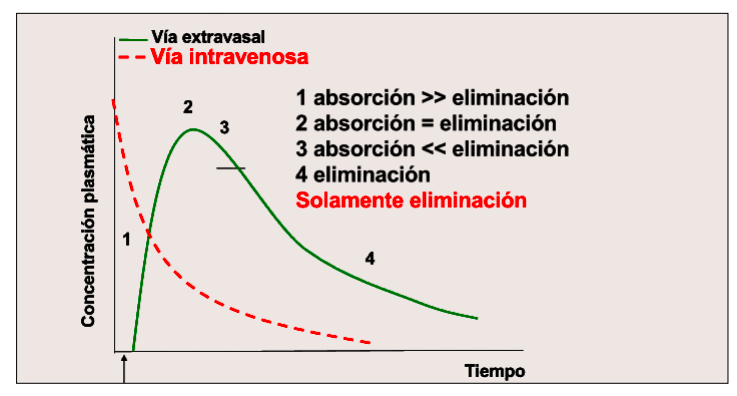
Izquierda grafica de intravenosa no se puede medir la absorción, pasa directamente a la distribución por el metodo de administración.
Derecha intramuscular, si se absorbe, luego se distribuye
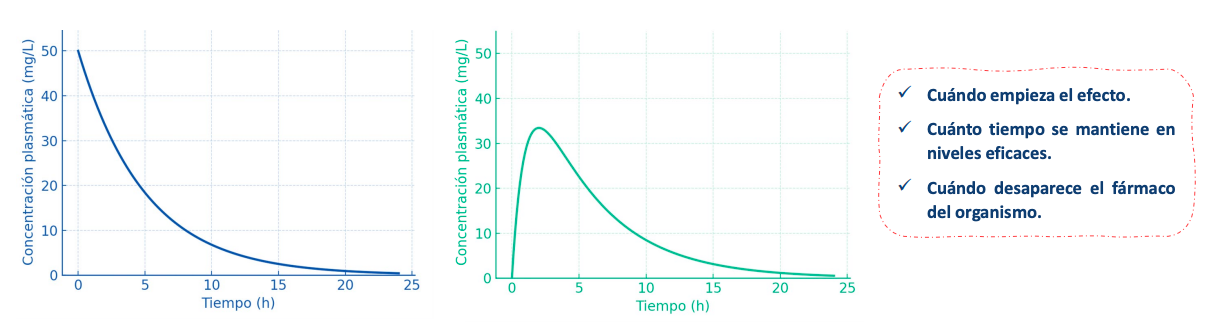
Con estas graficas y modelos podemos saber:
- Cuándo empieza el efecto.
- Cuánto tiempo se mantiene en niveles eficaces.
- Cuándo desaparece el fármaco del organismo.
IMPORTANTE Cuando la biodisponibilidad es el 100% la via intravenosa cortará al resto en su punto mas alto Cuando solo hay procesos de eliminación en las vias todas van a ser paralelas a la intravenosa
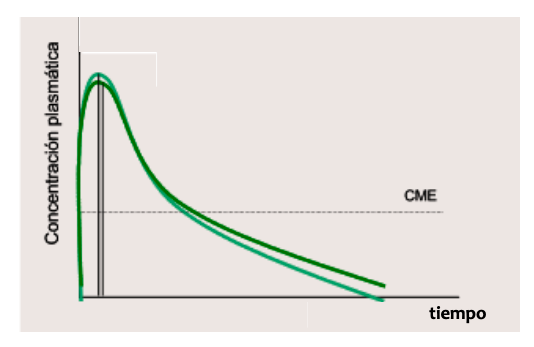
Partes de una gráfica:
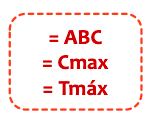
- Tmax o tiempo que pasa desde la administración al efecto maximo o Cmax
- Periodo de latencia, tiempo que pasa desde que se administra hasta el efecto o concentración minima eficaz
- Cmax o concentración maxima en sangre (punto mas alto)
- CME o concentración minima eficaz
- CMT o concentración minima tóxica (ha de estar siempre por debajo de la Cmax)
- Rango terapeutico: el espacio entre CME y CMT
Los modelos farmacocinéticos
La farmacocinética considera al organismo dividido en compartimientos, acuosos o no, que se definen como sectores reales o virtuales del organismo al cual puede acceder o del cual puede salir un fármaco.
Hay dos modelos, monocompartimental y bicompartimental. Los modelos bicompartimental son muy complejos así que solo nos centraremos en el monocompartimental que estudia los efectos como si el cuerpo fuera un solo compartimento y todo se distribuyese de forma homogenea.
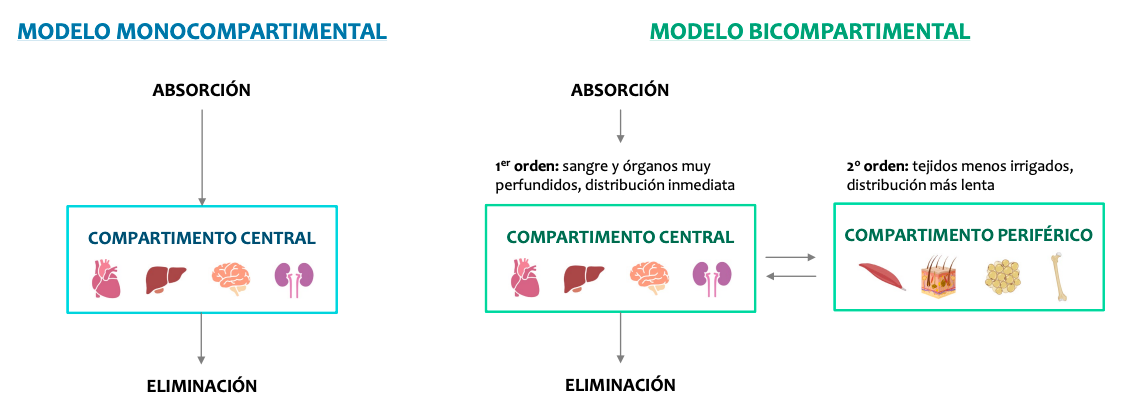
El modelo monocompartimental
- Es el más sencillo: el organismo se considera un único compartimento homogéneo.
- El fármaco, tras su administración, se distribuye rápida y uniformemente.
- La concentración plasmática desciende de manera exponencial según se va eliminando.
- Es útil para la mayoría de los fármacos de uso común, y como punto de partida para entender modelos más complejos.
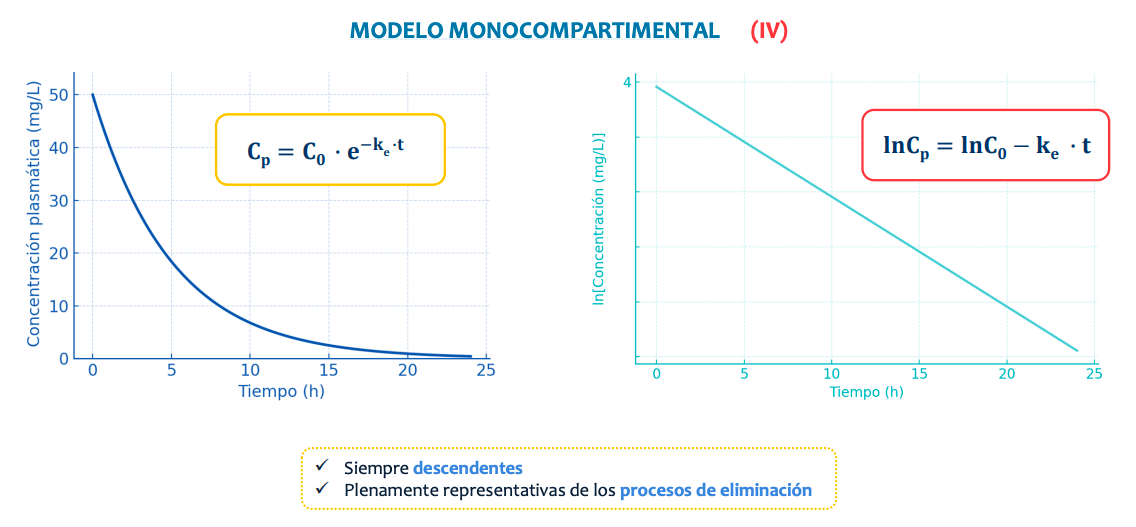
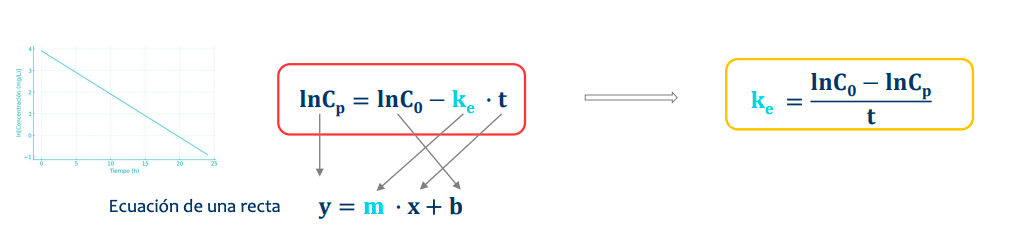
De manera natural la representación sigue una curva pero, aplicando un logaritmo la transformamos en una recta, simplificando el proceso y así pudiendo saber:
- La constante de velocidad de eliminación o ke
- Tiempo de vida media (t1/2)
- Volumen de distribución (Vd)
- Seran siempre descendientes al usar como base la concentración en via intravenosa y representarán solo el proceso de eliminación
Constante de velocidad de eliminación (ke)
- Indica la fracción del fármaco que se elimina por unidad de tiempo.
- Se mide en tiempo⁻¹ (ej. h⁻¹) (tiempo recíproco).
- Relaciona la concentración remanente en un tiempo dado con la que existe en la unidad de tiempo inmediatamente anterior.
- Es igual a la ecuación de la recta, despejamos para estudiar la ke (o la pendiente de la recta)
- IMPORTANTE, independientemente de la via de administración la ke o constante de velocidad de eliminación es especificia del farmaco y siempre será la misma
Tiempo de vida media (t1/2)
- Tiempo necesario para que la concentración plasmática de fármaco se reduzca a la mitad.
- Se expresa en unidades de tiempo real no en tiempo recíproco como la constante de eliminación.
- Aplicación clínica -> establecimiento de pautas posológicas

“Un perro recibe una dosis de un fármaco y alcanza una concentración plasmática de 16 µg/ml. La concentración mínima eficaz (CME) es de 2 µg/ml y la vida media (t½) del fármaco es de 4 horas. ¿Cuántas horas después de la administración volveríais a dar la siguiente dosis para mantener la eficacia del tratamiento?”
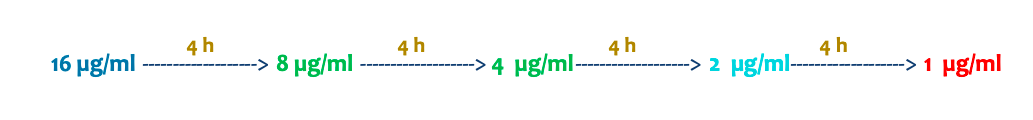
La respuesta es que habría que administrarlo a las 12 horas para evitar que la dosis baje de 2micro gramos/ml y no por encima de la concentración minima toxica.
Factores que modifican la vida media
- Dosis y coadministración de fármacos
- Algunos fármacos inducen o inhiben enzimas hepáticas.
- pH urinario
- Ácidos débiles → se eliminan más si la orina es básica (se ionizan más).
- Bases débiles → se eliminan más si la orina es ácida (se ionizan más).
- Especie, raza y edad
- Gatos (aspirina), Collies (mutación MDR1), jóvenes o geriátricos.
- Patologías
- Insuficiencia hepática, insuficiencia renal-> vida media más larga.
- Deshidratación (aclaramiento plasmático más lento) -> vida media más larga.
Página 15 - Clase del 2/10
Volumen de distribución - Modelo Monocompartimental (IV)
- Es una constante de proporcionalidad que relaciona la cantidad de fármaco existente en el organismo en un instante dado con su concentración plasmática en el mismo instante. Cuando analizamos un farmaco en una muestra de sangre la concentración sera igual en el resto del partes (Si la distribución es homogénea e instantanea)
- Unidades: litros ó litros/kg p.v.
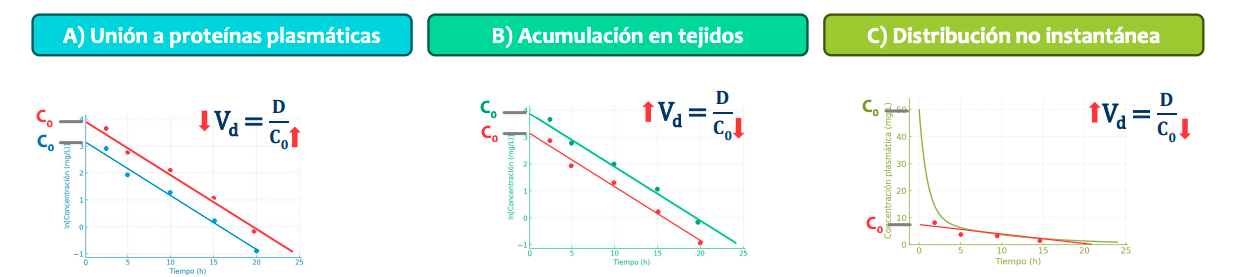
Está falseado (es decir, que el dato que da no es el correcto) si hay:
-
Unión a proteinas plasmaticas. Hay proporción libre (que es la que pasa a organos) y unida a proteinas, si la fración unida es muy grande el volumen de distribución no se ajustará a la realidad. La linea roja es la correcta, la concentración o C0 es mayor de la que aparentemente nos dan las pruebas, por tanto el volumen de distribución será menor del que vemos
-
Acumulación en tejidos: El volumen de distribución es mayor que el aparente y la concentración menor de la aparente. Como se acumula en organos la concentración real es menor que la que que el organismo nos dice ya que se acumula
-
Distribución no instantanea: Al principio se distribuye mas rapido y luego se estabiliza y baja la velocidad. La concentración real es menor y el volumen de distribución real es mayor, esto es así porque nos perdemos la fase de pico al ser tan alto.
REVISAR, PEDIR A ALGUIEN QUE LO EXPLIQUE
Compartimentos en el organismo:
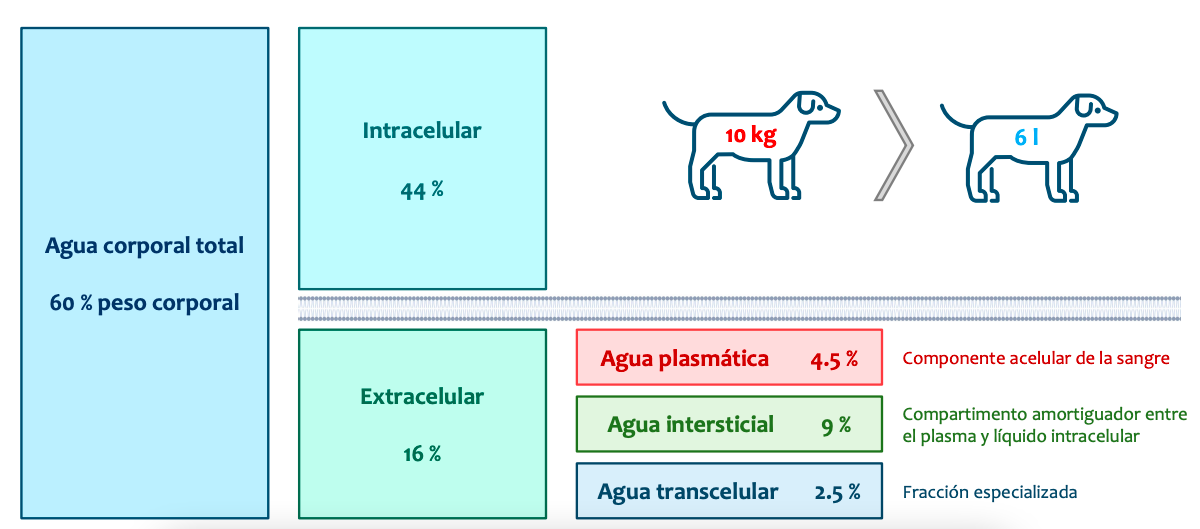
Puede entrar preguntas de calcular un volumen de distribución y explicarlo. Se administra a un gato un fármaco a una dosis de 20 mg/kg. La concentración plasmática inicial es de 34 mg/l. ¿Cuál es el volumen de distribución de este fármaco y cómo lo interpretas?
 Al ser un 60% si revisamos los compartimentos vemos que esto significa que se distribuirá por todos los compartimentos, porque hay un 60% del peso corporal en agua, si se pasa del 60%
Al ser un 60% si revisamos los compartimentos vemos que esto significa que se distribuirá por todos los compartimentos, porque hay un 60% del peso corporal en agua, si se pasa del 60%
Aclaramiento plasmático (Cl) - Modelo Monocompartimental (IV)
Es el volumen de plasma depurado de fármaco por unidad de tiempo por el conjunto de los órganos eliminadores del organismo.
-
Unidades: volumen/tiempo
-
Relaciona la capacidad de eliminación con el volumen donde está distribuido el fármaco.
-
Es una fórmula teórica, matemática, derivada del modelo monocompartimental.
-
Es la deducción matemática a partir de cómo cae la concentración plasmática con el tiempo (curvas plasmáticas).
-
Mide directamente qué volumen de plasma queda “limpio” de fármaco por minuto en función de lo que aparece en orina.
-
Es una fórmula práctica y experimental, útil en estudios de balance.
-
Mide experimentalmente lo que sale por orina en relación con lo que había en plasma.

Mientras se absorbe se elimina, en todo momento todas las acciones ocurren a la vez (Absorción, distribución, eliminación)
Vias extravasculares:
- 𝑒𝑒−𝐾𝐾𝑎𝑎𝑡𝑡 refleja la absorción.
- 𝑒𝑒−𝐾𝐾e𝑡𝑡 refleja la eliminación.
- La curva resultante es ascendente hasta alcanzar Cmáx (cuando la velocidad de absorción = velocidad de eliminación) y luego descendente.
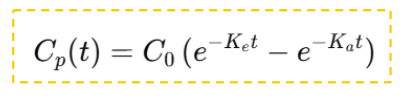
Constante de velocidad de absorción (ka)
- La concentración de fármaco que se absorbe por unidad de tiempo va disminuyendo exponencialmente a medida que disminuye la concentración del fármaco en el compartimento del que desaparece.
- Se mide en tiempo⁻¹ (ej. h⁻¹) (tiempo recíproco).
- Ejemplo: ka = 0,05 h-1 indica que en 1 hora se absorberá el 5 % de las moléculas del fármaco en disolución.
Semivida de absorción (t1/2a)
- Tiempo que tarda en reducirse a la mitad el número de moléculas de fármaco que quedan por absorberse.
¿Cómo se calcula la constante de velocidad de absorción (ka)?
Métodos directos
Métodos indirectos Método de los residuales
- Fundamento: restar del nivel plasmático teórico que existiría de no haber mediado absorción, el valor obtenido experimentalmente en el mismo instante. Esta diferencia, representa la fracción que no ha pasado al plasma y permanece en el lugar de absorción.
Realización:
- Extrapolar la fracción recta de la representación semilogarítmica de las curvas de nivel plasmático.
- Calcular las diferencias entre las concentraciones extrapoladas y los valores experimentales.
- Representar las diferencias obtenidas en la gráfica.
- Unir dichas diferencias mediante una recta cuya pendiente será ka.
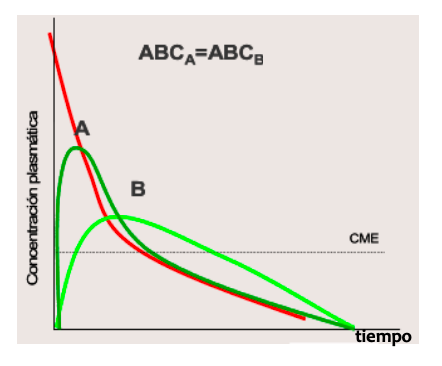
Biodisponibilidad (F)
- Es la cantidad de fármaco absorbida en relación a la cantidad de fármaco administrada.
- Se calcula el área bajo la curva (ABC) de Cp del fármaco: ABC = C0/ke
- Se expresa en % en relación a la vía intravascular: biodisponibilidad del 100 %.
- Para el mismo principio activo: comparamos el ABC por diferentes vías de administración y formas farmacéuticas con ABCiv = 100 %, y nos permite determinar la dosis adecuada.
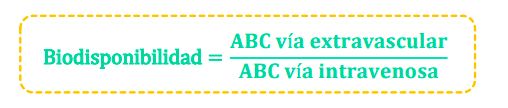
Bioequivalencia
- Parámetro más restrictivo.
- Requisito para especialidades farmacéuticas genéricas (EFG).
- Un fármaco genérico, además de la misma formulación y principio activo que otro fármaco ya comercializado ha de ser bioequivalente:
- diferencias aceptables ± 20%.
Influencia en la vía de administración
- El tramo descendente exponencial tiene la misma forma en todas las curvas (ke y t1/2 mismo valor para todas).
- El tramo exponencial se desplaza a la derecha a medida que la aparición del fármaco en sangre se hace más lenta (el fármaco permanece más en el plasma cuanto más lenta es su absorción).
- El valor de la intersección de la recta de la fase terminal extrapolada en el eje de ordenadas es mayor cuanto más lenta es la absorción (el antilog de C0 aumenta a medida que Ka disminuye). Las rectas de pendiente Ke son paralelas.
- El valor máximo de Cmax es más bajo y la curva es más roma cuanto más lenta es la absorción.
- La curva IV corta a todas en sus máximos. Absorción completa y dosis iguales.
- El valor de ABC de niveles plasmáticos es constante, sea cual fuere la administración utilizada.
- Cuando usamos los logaritmos para sacar la recta, es mayor cuanto mas lenta es la absorción y todas son paralelas
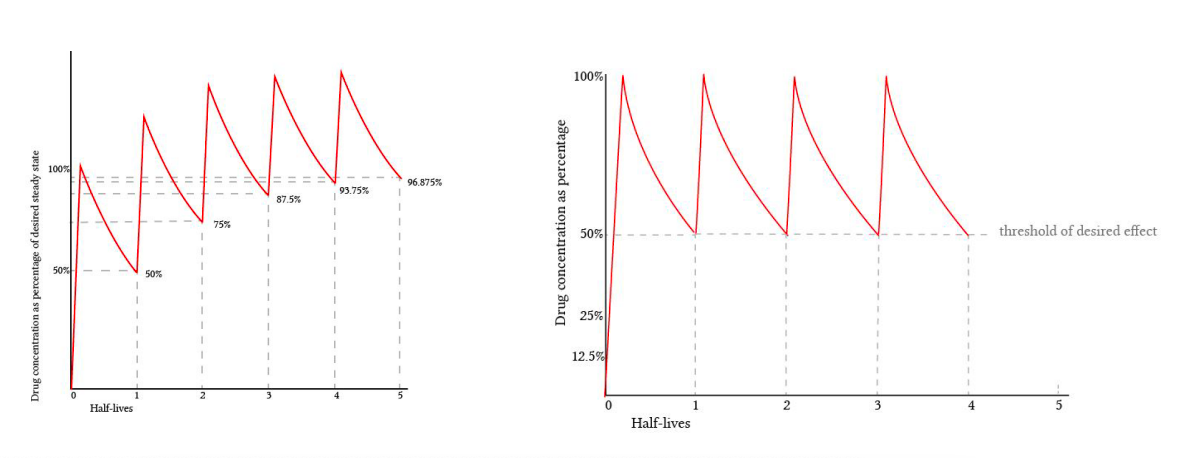
Pautas de dosificación
Regimen de dosificación racional
Pautas de dosificación: Es la pauta de administración de medicamento mediante la cual se alcanza un nivel terapeútico eficaz en la biofase, se mantiene éste durante todo el tratamiento y se previene la acumulación del medicamento en el organismo.
Parámetros fundamentales en un régimen de dosificación
- C min: Concentración plasmática mínima eficaz
- C max: Concentración plasmática, inferior a la concentración plasmática tóxica
- D: Dosis de mantenimiento sensiblemente igual a la dosis mínima eficaz, repetida a intervalos fijos
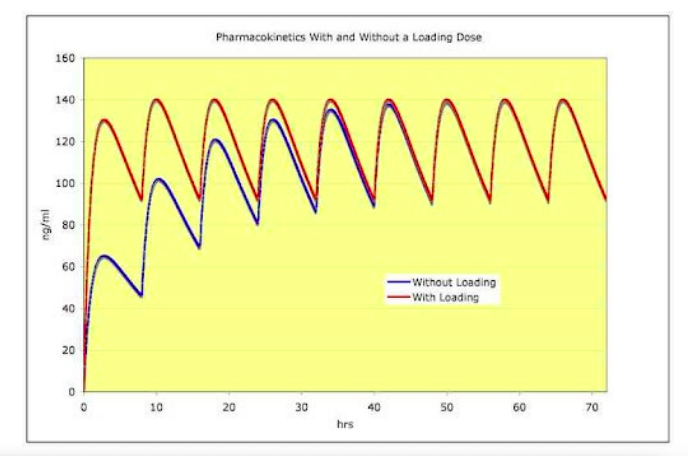
- D*: Dosis de choque inicial: es el doble de la de mantenimiento, pero solo se utiliza para la primera vez.
- 𝑻𝑻: Intervalo de dosificación o tiempo transcurrido entre dos administraciones
Para esto se usa la dosis de choque inicial, para llegar a la estabilidad más rapido:
Dosis de mantenimiento vía oral
D: dosis 𝑻𝑻: Intervalo de dosificación o tiempo transcurrido entre dos administraciones. D/ 𝑻𝑻 : tasa de dosis por intervalo de administración (dosis/horas; dosis/días) Cmss: Concentración del fármaco media deseada en equilibrio estacionario. F: biodisponibilidad del fármaco por vía oral. Cl: aclaramiento del fármaco
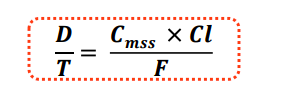 Con esta fórmula establecemos la dosis/tiempo
Con esta fórmula establecemos la dosis/tiempo
Queremos alcanzar una concentración plasmática (Cp) objetivo de 6 mg/l para un fármaco cuyo rango terapéutico es de 4 a 10 µg/ml. Se conocen los siguientes parámetros: aclaramiento (Cl): 25 l/h; biodisponibilidad (F): 83%; t1/2 = 4 h.
Pero necesitamos fraccionar la dosis estableciendo una pauta de dosificación para que las Cp del fármaco (tanto la Cmax como la Cmin) estén siempre dentro del rango terapéutico del fármaco: Cuando T = t1/2 Cmax/Cmin ≈ 2

Repasar
No comments to display
No comments to display