Tema 8 - Farmacocinética Cuantitativa - 1/10
Clase del 1/10
Presentación
Preguntas a responder:
¿A qué velocidad se absorbe un fármaco después de administrarlo?
¿Qué concentración alcanzará en plasma y durante cuánto tiempo se mantendrá en un nivel terapéutico?
¿Con qué intervalo debemos administrar la medicación para que sea eficaz y segura?
¿Cuál es la dosis óptima para una especie determinada?
La farmacocinética cuantitativa nos permite, mediante modelos matemáticos sencillos, conocer los siguientes parámetros farmacocinéticos:
Velocidad de absorción
Cantidad de fármaco absorbido
Modo de distribución del fármaco
Velocidad de eliminación
…
Establecer un régimen posológico:
Dosis óptima
Intervalo de administración
Duración del tratamiento
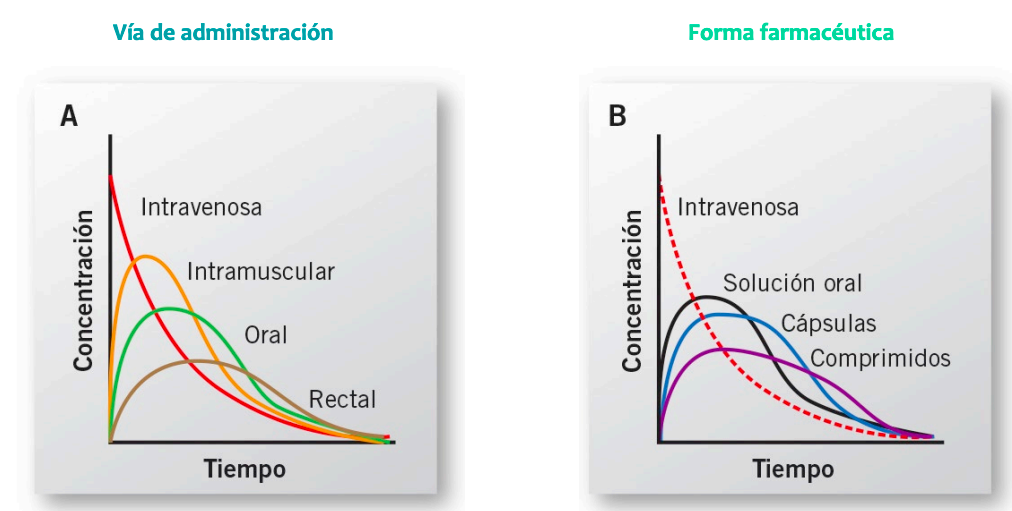
Izquierda grafica de intravenosa no se puede medir la absorción, pasa directamente a la distribución por el metodo de administración.
Derecha intramuscular, si se absorbe, luego se distribuye
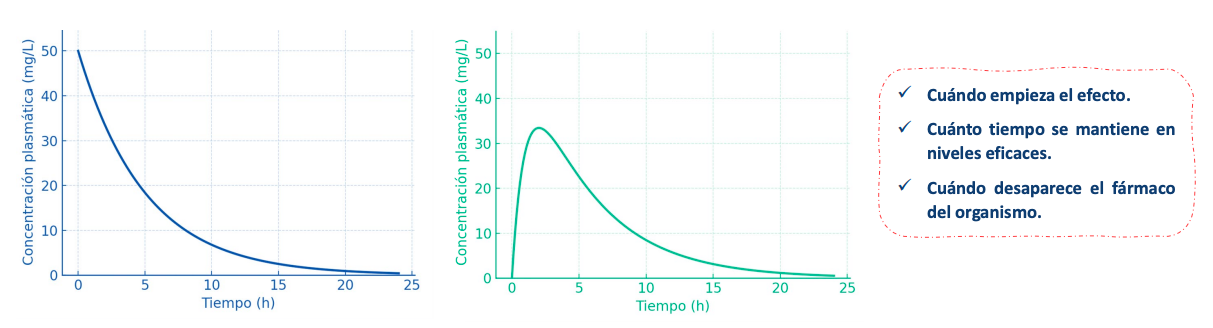
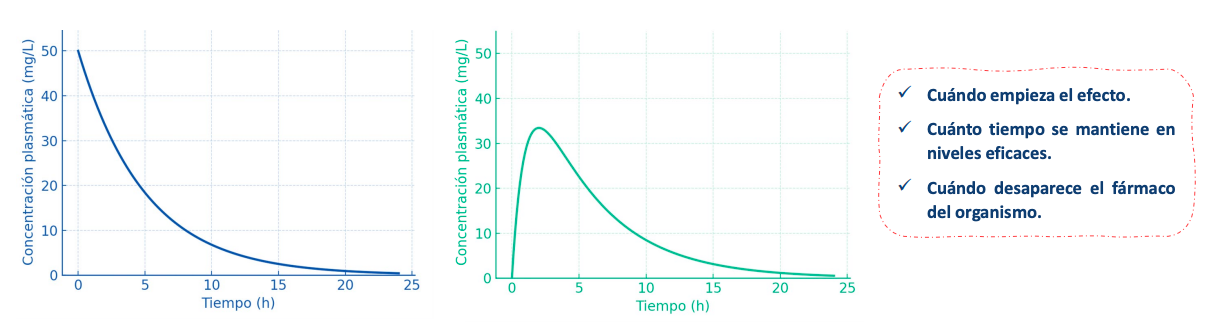
Con estas graficas y modelos podemos saber:
Cuándo empieza el efecto.
Cuánto tiempo se mantiene en niveles eficaces.
Cuándo desaparece el fármaco del organismo.
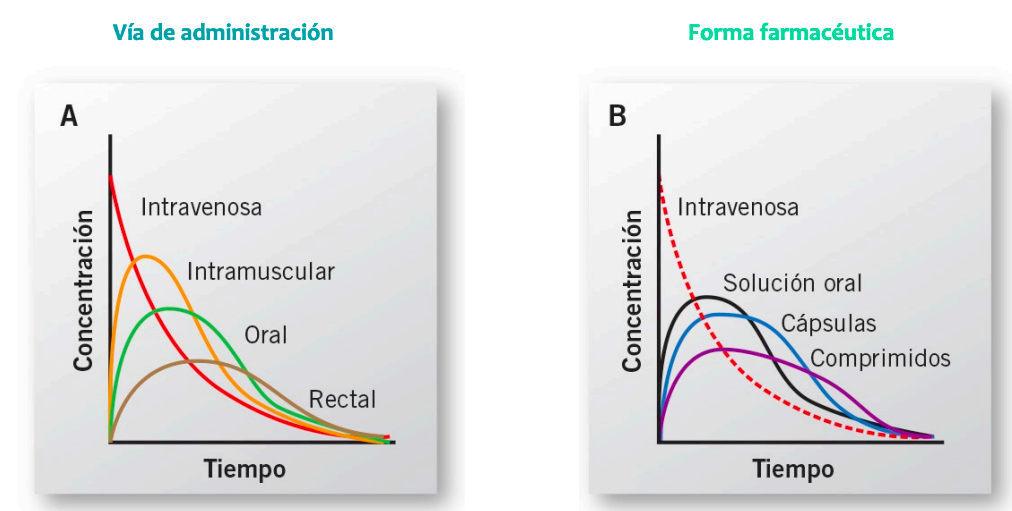
IMPORTANTE Cuando la biodisponibilidad es el 100% la via intravenosa cortará al resto en su punto mas alto
Cuando solo hay procesos de eliminación en las vias todas van a ser paralelas a la intravenosa
Partes de una gráfica:
Tmax o tiempo que pasa desde la administración al efecto maximo o Cmax
Periodo de latencia, tiempo que pasa desde que se administra hasta el efecto o concentración minima eficaz
Cmax o concentración maxima en sangre (punto mas alto)
CME o concentración minima eficaz
CMT o concentración minima tóxica (ha de estar siempre por debajo de la Cmax)
Rango terapeutico: el espacio entre CME y CMT
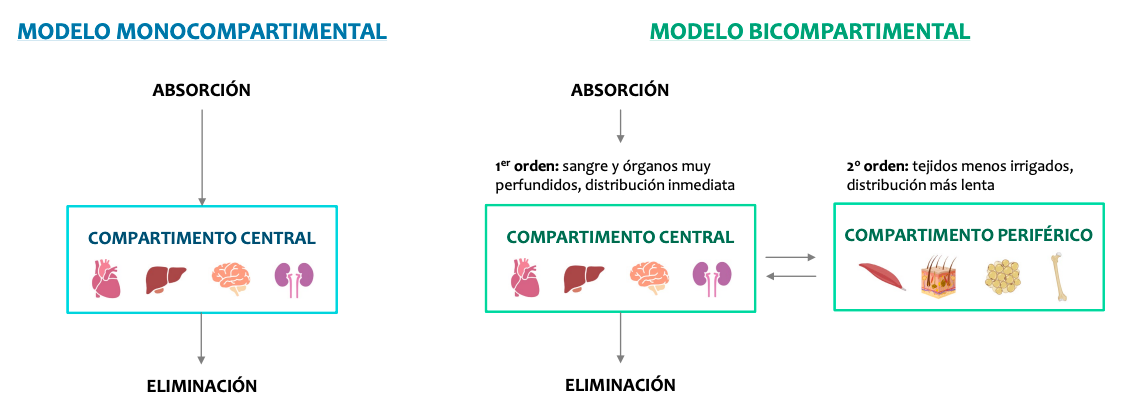
Los modelos farmacocinéticos
La farmacocinética considera al organismo dividido en compartimientos, acuosos o no, que se definen como sectores reales o virtuales del organismo al cual puede acceder o del cual puede salir un fármaco.
Hay dos modelos, monocompartimental y bicompartimental.
Los modelos bicompartimental son muy complejos así que solo nos centraremos en el monocompartimental que estudia los efectos como si el cuerpo fuera un solo compartimento y todo se distribuyese de forma homogenea.
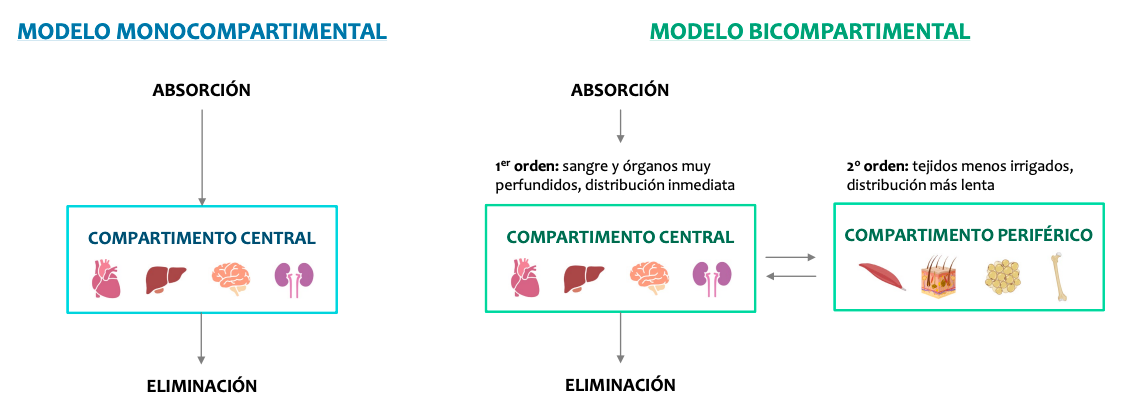
El modelo monocompartimental
Es el más sencillo: el organismo se considera un único compartimento homogéneo.
El fármaco, tras su administración, se distribuye rápida y uniformemente.
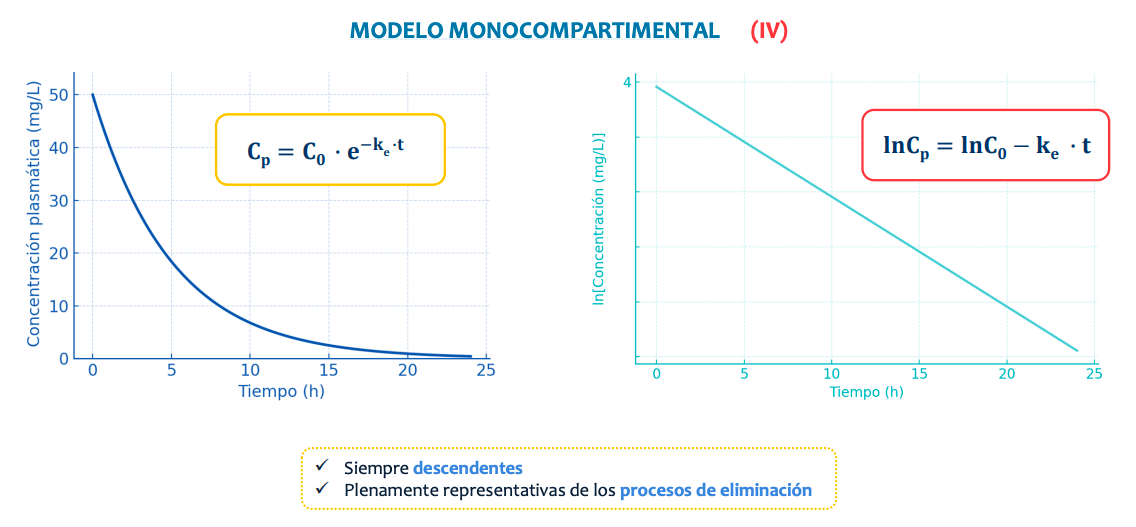
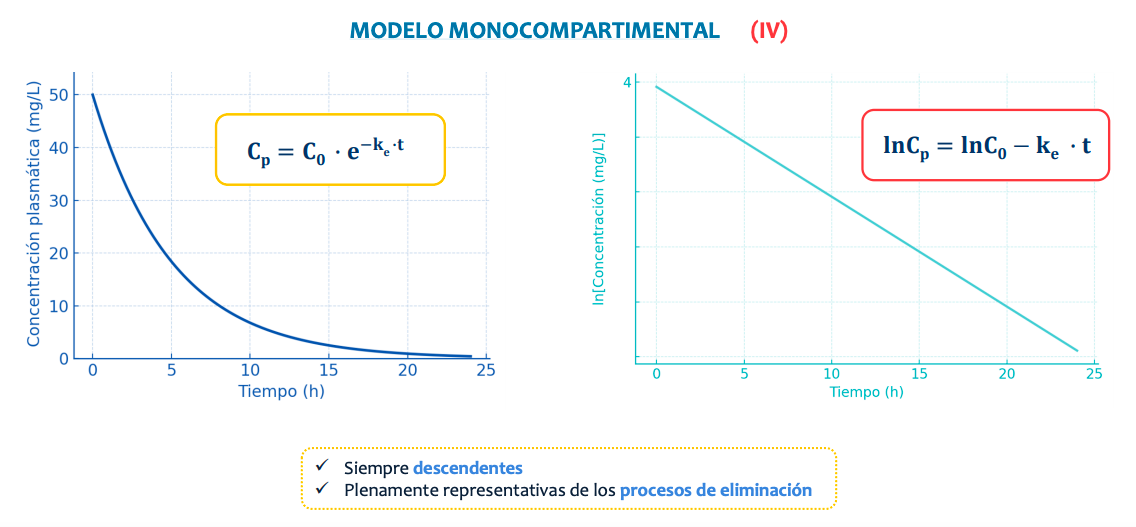
La concentración plasmática desciende de manera exponencial según se va eliminando.
Es útil para la mayoría de los fármacos de uso común, y como punto de partida para entender modelos más complejos.
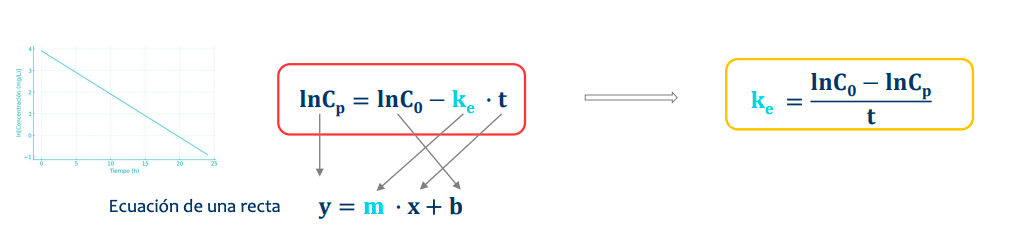
De manera natural la representación sigue una curva pero, aplicando un logaritmo la transformamos en una recta, simplificando el proceso y así pudiendo saber:
La constante de velocidad de eliminación o ke
Tiempo de vida media (t1/2)
Volumen de distribución (Vd)
Seran siempre descendientes al usar como base la concentración en via intravenosa y representarán solo el proceso de eliminación
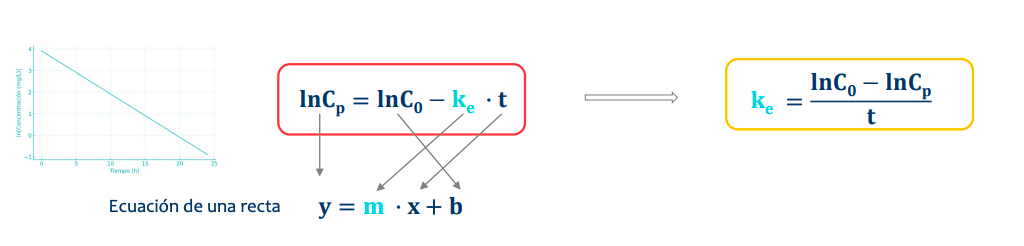
Constante de velocidad de eliminación (ke)
Indica la fracción del fármaco que se elimina por unidad de tiempo.
Se mide en tiempo⁻¹ (ej. h⁻¹) (tiempo recíproco).
Relaciona la concentración remanente en un tiempo dado con la que existe en la unidad de tiempo inmediatamente anterior.
Es igual a la ecuación de la recta, despejamos para estudiar la ke (o la pendiente de la recta)
IMPORTANTE, independientemente de la via de administración la ke o constante de velocidad de eliminación es especificia del farmaco y siempre será la misma
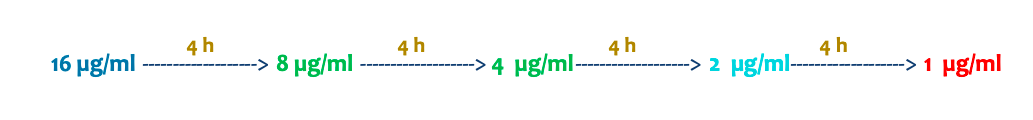
Tiempo necesario para que la concentración plasmática de fármaco se reduzca a la mitad.
Se expresa en unidades de tiempo real no en tiempo recíproco como la constante de eliminación.
Aplicación clínica -> establecimiento de pautas posológicas

“Un perro recibe una dosis de un fármaco y alcanza una concentración plasmática de 16 µg/ml. La concentración mínima eficaz (CME) es de 2 µg/ml y la vida media (t½) del fármaco es de 4 horas. ¿Cuántas horas después de la administración volveríais a dar la siguiente dosis para mantener la eficacia del tratamiento?”
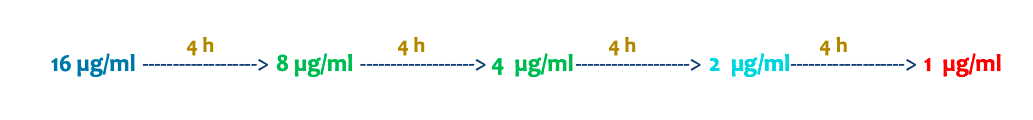
La respuesta es que habría que administrarlo a las 12 horas para evitar que la dosis baje de 2micro gramos/ml y no por encima de la concentración minima toxica.
Factores que modifican la vida media
Dosis y coadministración de fármacos
Algunos fármacos inducen o inhiben enzimas hepáticas.
pH urinario
Ácidos débiles → se eliminan más si la orina es básica (se ionizan más).
Bases débiles → se eliminan más si la orina es ácida (se ionizan más).
Especie, raza y edad
Gatos (aspirina), Collies (mutación MDR1), jóvenes o geriátricos.
Patologías
Insuficiencia hepática, insuficiencia renal-> vida media más larga.
Deshidratación (aclaramiento plasmático más lento) -> vida media más larga.
Página 15 - Clase del 2/10