Tema 5 - Farmacocinética: Distribución - 17/09
Clase del 17/09 dada por Laura Presentación
Farmacocinetica
RELLENAR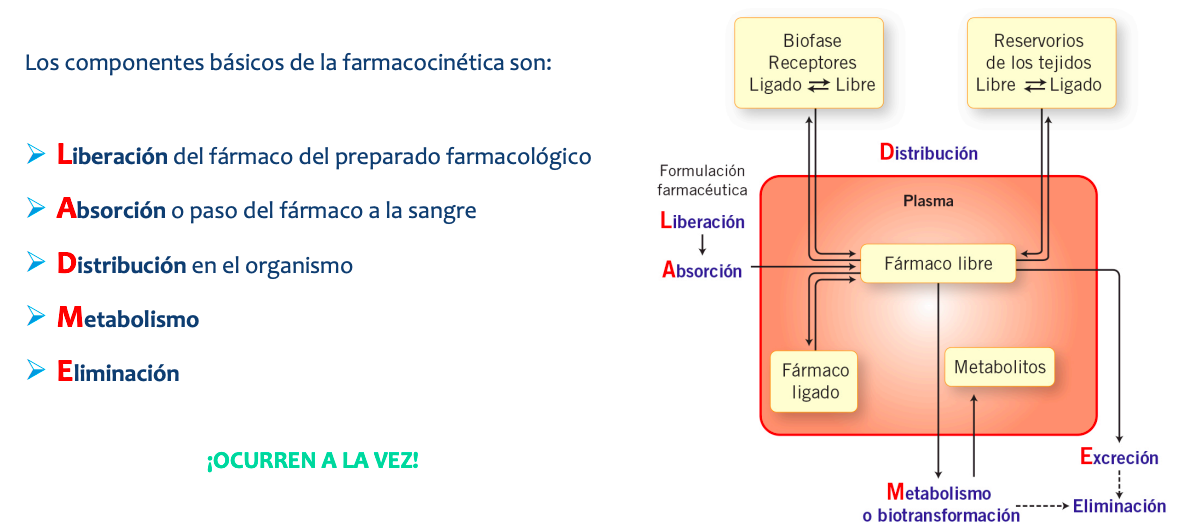
Distribución
La distribución es el proceso mediante el cual el fármaco se incorpora desde la circulación sanguínea hacia los distintos órganos y tejidos corporales, pasando a través de diversas membranas biológicas.
PaginaLas 7moleculas en la sangre pueden ir:

En cuanto a la fracción unida a proteinas:

¿Por qué es importante la fracción ligada a proteínas plasmáticas?
¿Qué factores que alteran la unión a proteínas plasmáticas?
¿Cuándo tiene importancia clínica?
No todos los farmacos a los mismos puntos de las proteinas.
Tiene importancia cuando dos farmacos:
- Si afectan a la misma proteína que fija al fármaco.
- Si afecta al mismo lugar de fijación.
- Si el fármaco se une más del 80 % a las proteínas plasmáticas.
Salida de los capilares: Difusion pasiva o filtración por poros llenos de agua. Liposolubles por membrana lipidica, hidrosolubles por poros con agua pero generalmente son liposolubles.
La velocidad de salida depende de:
- Naturaleza del fármaco (liposoluble generalmente mas rapido)
- Tamaño (mas lento)
- Coeficiente de partición
- Unión a proteínas plasmáticas (mas lento)
Distribución de los liquidos en el organismo
En los tejidos los fármacos están disueltos en el líquido intersticial y en el líquido intracelular.
Alcanzarán distintas concentraciones en unas u otras áreas del organismo dependiendo de:
- Flujo sanguíneo regional
- Especial afinidad por un tejido donde se acumula
- Masa de los distintos órganos en relación con el peso corporal
- pH de los líquidos corporales
- Liposolubilidad y grado de ionización
- SOLO ES EL FARMACO LIBRE EL QUE PASARA A TEJIDOS, EL UNIDO A PROTEINAS NO
Agua corporal total, 60% del peso corporal, 44% intracelular y 16% extracelular (4,5% plasmatico, 9% intesticial, 2,5 transcelular
Es el volumen del líquido en el cual se encuentra distribuido o contiene el fármaco, este es aparente y no es real. Permite predecir la distribución de un fármaco en el organismo. Si es alto el Vd o Volumen de distribución es que penetra en todos los compartimentos, la concentracion del farmaco en tejidos sera alta

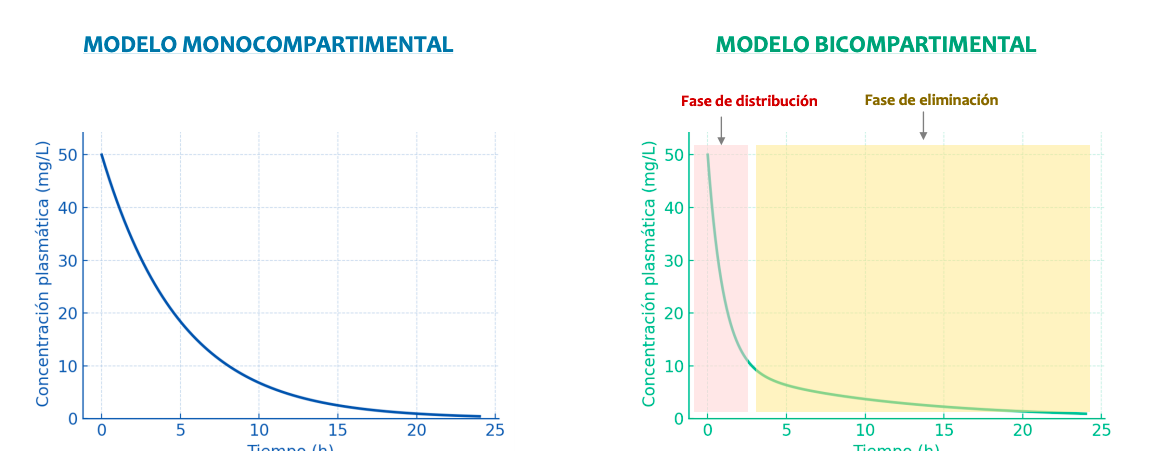
Modelos de distribución:
La farmacocinética considera al organismo dividido en compartimientos, acuosos o no, que se definen como sectores reales o virtuales del organismo al cual puede acceder o del cual puede salir un fármaco. Hay dos modelos:
- Modelo Monocompartimental: Absorción y distribución inmediata. Usaremos el modelo monocompartimental en practicas y examen
- Modelo Bicompartimental, 1er orden: Absorción de los organos mas perfundidos, distribución inmediata y luego un 2o orden: tejidos menos irrigados, distribucion mas lenta
Curva de concentración plasmatica. No es homogenea en la bicompartimental
Factores que modifican la distribución:
- Propiedades fisicoquímicas del fármaco: se distribuyen mejor los medicamentos mas liposolubles, no ionizados y de bajo PM.
- Flujo sanguíneo del tejido: los fármacos alcanzan concentraciones más elevadas y con mayor rapidez en los órganos mejor perfundidos.
- Afinidad del fármaco por el tejido: por ejemplo, los digitálicos tienen afinidad por el tejido cardiaco y las tetraciclinas por el hueso.
- Contenido lipídico del tejido: existen fármacos muy liposolubles que se acumulan en la grasa quedando atrapados a ese nivel y son liberados con lentitud.
- Grado de unión a las proteínas plasmáticas: el grado de unión entre el fármaco y la proteína plasmática depende de: Naturaleza del fármaco, Afinidad del fármaco para la proteína plasmática, Disponibilidad de proteínas plasmáticas
Barreras corporales especiales: ofrecen dificultad al pasaje de algunos fármacos.
Barrera hematoencefálica (BHE):
- Solo es permeable a sustancias liposolubles.
- Las sustancias muy ionizadas como las aminas cuaternarias o las penicilinas normalmente son incapaces de atravesarlas. (en situacion fisiologica, si esta inflamado como en meningitis puede pasar)
- Células epiteliales (vasos capilares SNC) muy unidas, no hay poros acuosos entre las células; impide difusión de sustancias polares de bajo PM.
- Células gliales (astrocitos) rodean los capilares del SNC.
Barrera hemato-placentaria:
- Este órgano de intercambio materno-fetal consta de 3 estratos de tejidos fetales
- La penetración es mínima para los fármacos con alto grado de disociación o baja liposolubilidad.
- Los amonios cuaternarios y las sustancias hidrosolubles de PM superior a 1.000 no atraviesan la BP; sin embargo, la placenta es permeable a fracciones no ionizadas, a los no electrolitos liposolubles (éter, cloroformo), a las hormonas esteroideas, salicilatos, atropina, barbitúricos, antibióticos, alcaloides, etc.
- Los fármacos la atraviesan por difusión pasiva, facilitada y pinocitosis.
¿Por qué hay diferencias en la distribución de un fármaco entre especies?
- Composición corporal (porcentaje de grasa y agua).
- Flujo sanguíneo y perfusión tisular.
- Niveles y tipos de proteínas plasmáticas.
- Permeabilidad de barreras biológicas (BHE, placenta).
- Afinidad tisular y actividad metabólica.
Importante:
- La distribución es clave para comprender eficacia y toxicidad.
- No solo importa la dosis, sino a dónde llega el fármaco.