Conceptos
Farmacología general
Tema 2 - Introducción
Farmacología veterinaria: Es la rama de las ciencias biomédicas que estudia las propiedades, efectos y mecanismos de acción de los fármacos en animales, tanto domésticos como silvestres. Incluye también la aplicación clínica de dichos conocimientos para la prevención, diagnóstico, tratamiento y control de enfermedades.
| Concepto | Definición / Características |
|---|---|
| Fármaco | Sustancia química de estructura conocida que produce un efecto biológico al administrarse a un ser vivo. Molécula bioactiva que interactúa con receptores u otras macromoléculas para dar lugar a una ccion y efecto |
| Medicamento | Preparación química que contiene uno o más fármacos (principios activos) + excipientes. Se administra con el fin de provocar un efecto terapéutico, preventivo diagnostico o correctivo. NUNCA CREA FUNCION NUEVA |
| Principio activo | Sustancia o combinación de sustancias con propiedades curativas, preventivas o que modifican funciones fisiológicas mediante acción farmacológica, inmunológica o metabólica. |
| Forma farmacéutica | Disposición en la que se presentan los principios activos y excipientes para constituir un medicamento. Incluye la presentación y vía de administración (ej. comprimidos, soluciones, inyectables). |
| Excipiente | Sustancia inactiva que acompaña al principio activo en el medicamento. Facilita la preparación, conservación, administración y estabilidad del producto. |
| Adyuvante | Sustancia añadida para potenciar o mejorar la acción del principio activo, sin tener efecto terapéutico propio. Se usa en vacunas y formulaciones específicas. |
Definiciones de especialidades dentro de la Farmacoloía
Farmacocinetica: Estudia la evolución temporal de las concentraciones farmacológicas alcanzadas en diferentes regiones del organismo durante y después de la administración. “Lo que el organismo hace con el fármaco”. Estudia de forma dinamica y cuantitativa el LADME, Liberación, Absorción, Distribucion, Metabolismo y Eliminación del farmaco.
En resumen, la evolución de la concentración del farmaco en el organismo
Farmacodinamia: Estudia los acontecimientos derivados de la interacción entre el fármaco y su receptor u otro lugar primario de acción. “Lo que el fármaco hace al organismo”. Efectos bioquimicos, fisiologicos de los medicamentos, su mecanismo de accion, correlacion, efectos y estructura.
En resumen, estudia la relación concentración / efecto = resultado clinico
Farmacoterapia: Es el tratamiento de las enfermedades mediante medicamentos. La farmacoterapia terapeutica es la que estudia la curacion o alivio de enfermedades.
Farmacovigilancia: ciencia y las actividades vinculadas a la detección, evaluación, comprensión y prevención de las sospechas de acontecimientos adversos o cualquier otro problema relacionado con un medicamento veterinario.
Farmacognosia: Describe las drogas o medicamentos considerando su origen, características organolépticas, físicas y químicas.Estudia los principios activos de origen natural que pueden poseer un potencial terapéutica o aplicación en la industria
Farmacotecnia: Es la técnica de transformación de un principio activo en un medicamento apto para su uso. Entiende de la conveniente preparación de los medicamentos para su utilización terapéutica.
Farmacogenética: Estudio de las influencias genéticas en la respuesta a los fármacos. FARMACOGENÓMICA. Describe el uso de la información genética para elegir el tratamiento farmacológico más indicado en cada caso.
Farmacoeconomía: Cuantifica en términos económicos los costes y los beneficios de los fármacos para usos terapéuticos.
Farmacoepidemiología: Estudio de los efectos de los fármacos en la población.
Farmacopatología: Estudia los efectos secundarios de los fármacos en el organismo.
Tema 3 - Farmacocinética - Liberación
Farmacocinetica: la evolución de la concentración del farmaco en el organismo
Estudio del comportamiento o evolución temporal, desde un punto de vista cuantitativo, de los fármacos en el organismo, desde que son administrados hasta que se eliminan.
Componentes basicos de la farmacocinetica:
- Liberación del farmaco del preparado farmacológico
- Absorción o paso del farmaco a la sangre
- Distribución por el organismo
- Metabolización (Generalmente hepatica)
- Eliminación (Generalmente renal)
IMPORTANTE, TODAS LAS FASES DE LA FARMACOCINETICA OCURREN A LA VEZ
| Parámetro | Parte del LADME relacionada | Explicación breve |
|---|---|---|
| Velocidad de absorción del fármaco | Absorción | Indica qué tan rápido pasa el fármaco desde el sitio de administración a la sangre. |
| Biodisponibilidad | Absorción (y parcialmente Metabolismo de primer paso) | Proporción del fármaco que alcanza la circulación sistémica en forma activa. |
| Vida media de eliminación | Eliminación (también influida por Metabolismo) | Tiempo necesario para reducir a la mitad la concentración plasmática del fármaco. |
| Concentración plasmática total | Resultado de Absorción + Distribución + Metabolismo + Eliminación | Refleja el equilibrio dinámico de todas las fases del LADME. |
| Área bajo la curva (AUC) | Absorción y Eliminación | Mide la exposición total del organismo al fármaco a lo largo del tiempo. |
¿Actúa igual un fármaco aplicado localmente que uno administrado por vía sistémica?
-
Local → El fármaco actúa directamente en el sitio de aplicación, con poca absorción sistémica y menos efectos generales. Mas concentración en el sitio de interes.
-
Sistémica → El fármaco pasa por todas las fases del LADME, alcanza la sangre y se distribuye por todo el organismo.
No actúan igual porque la farmacocinética cambia: local = efecto concentrado, sistémica = efecto global.
Liberación: Salida del farmaco de su forma farmaceutica que lo transporta, esto implica la disolución del farmaco en un medio corporal. Le influyen:
- Tamaño de la particula
- Solubilidad: Por ph del medio del farmaco (pKa) y formacion de sales o esteres
- Formulación del medicamento (diseño químico/tecnológico del medicamento.)
- Tecnica de elaboración
- Forma farmaceutica
Fases de la liberación:
- Disgregación: forma solida a particulas mas pequeñas = Solido grande a solido pequeño
- Disolución: forma solida pequeña a solución EL MAS IMPORTANTE = Solido pequeño a disolucion o liquido)
- Difusion: paso del farmaco disuelto por el fluido (No sanguineo, intra o extra celular)
Las fases de la liberación dependen mucho de la forma terapeutica, se libera mas rapido en formas farmaceuticas liquidas o solubles
Tema 4 - Farmacocinética - Absorción
La absorción es el paso del fármaco desde su lugar de administración al torrente sanguíneo A TRAVES DE LA MEMBRANA PLASMATICA.
La membrana plasmatica engloba a la celula y está formada por fosfolipidos y proteinas. Para que un farmaco atraviese la membrana ha de ser un farmaco NO DISOCIADO, es decir, LIPOSOLUBLE. Un farmaco disociado es hidrosoluble y será repelido por la membrana (Aun así pueden atravesarla en situaciones especiales y si tienen caracteristicas especiales).
Paso del farmaco a través de las membranas celulares:
Mecanismo de transporte pasivo o simple: A favor del gradiente de concentración sin gasto de energia y depende del peso molecular de la sustancia:
- Difusion simple (farmaco liposoluble no ionizado como el diacepam): Usado por el 80% de los farmacos. Permite el paso de un peso molecular de particulas de menos de 1000 daltons
- Filtración a traves de poros (hidrosolubles, Sodio o agua)
Mecanismo de transporte especializado :
- Difusion simple facilitada: A traves de proteinas transportadoras, TRANSPORTE PASIVO SATURABLE (Glucosa)
- Transporte activo (gasto de energia, Omeprazol)
Por incorporación con la membrana
- Pinocitosis
- Fagocitosis
Factores que determinan la difusion simple:
Liposolubilidad
- Directamente proporcional a la liposolubilidad del farmaco. Es decir, cuanto mas liposoluble, mas facil es que atraviesen la membrana por difusion simple
- Se determina mediante el coeficiente de partición lipido/agua mediante la disolucion de farmaco en solventes polares y apolares. IMPORTANTE, cuanto mas alto es el cociente de partición, mas liposoluble es el farmaco. (Al ser el lipido el numerador, un numero mas grande dara un mayor coeficiente). No confundir con coeficiente de disociación
Tamaño molecular:
- La velocidad de difusion es inversamente proporcional al tamaño molecular, es decir, cuanto menor tamaño, mas velocidad de difusion.
Ionización molecular:
- La mayoria de los farmacos son acidos o bases debiles que pueden estar o no ionizados. Cuando se ionizan se vuelven polares, hidrosolubles, la parte no ionizada es más liposoluble.
- La fraccion no ionizada determina la velocidad de pasaje a traves de las membranas. Directamente proporcional: cuanto mayor es la parte no ionizada, mayor es la velocidad de pasaje.
- Se puede calcular si conocemos si es un acido o base, el pKa del farmaco (Valor del pH en el cual un farmaco presenta un 50% de sus moleculas ionizadas y el otro no, si es un acido el numero estará por debajo de 7, si es una base estaraá por encima)
- pH del medio orgánico
Filtración a traves de poros:
- Consiste en el pasaje a favor de gradiente de los farmacos a traves de canales o poros de la membrana celular, depende del tamaño y el gradiente de concentración
- No necesita ATP
- Hay poros hidrofilos o canales acuosos
- Solo pasan moleculas pequeñas
- Del tamaño de los poros depende la velocidad de filtración
- SOLO PASAN MOLECULA NEUTRAS O NEGATIVAS ya que las proteinas tienen carga positiva
- Iones y agua es lo que mas pasa.
- El proceso de filtración glomerular es gracias a este proceso. Todos los farmacos libres con peso molecular menor a 70000 son filtrados al glomerulo
Difusion facilitada:
- No requiere gasto energetico
- Necesita una proteina transportdora
- Solo pasan modelas pequeñas apolares
- Lo usa la glucosa, aminoacidos, neurotransmisores...
Transporte activo
- En contra del gradiente de concentración (pasa del sitio con menos concentración al que mas)
- Requiere de transportadores especificos
- Es selectivo y saturable
- Requiere gasto de energia
Endocitosis y pinocitosis:
- Invaginacion de parte de lamembrana celular y atrapamiento de una particula solida o liquido para depositarla en el interior de la celula. La pinocitosis es la entrada de liquidos o disoluciones, fagocitosis es la inestion de particulas grandes o solidas como bacterias.
Factores que afectan a la absorción:

Grado de ionización y pH:
- Los fármacos son ácidos o bases débiles que en solución se disocian. Solo las moléculas no ionizadas son liposolubles. Solo las formas no ionizadas atraviesan membranas.
- La posibilidad de atravesar las membranas depende de la proporcion de moleculas no ionizadas y del pH del medio
- El grado de ionización se calcula por la constante de ionización o Ka y el pH del medio
- Se usa la ecuación de Henderson-Hasselbach

HA es un acido debil o porcion no disociada, A es el acido fuerte o disociado BOH es una base debil o porcion no disociada, B es la base fuerte o disociada
- Cuando el pH = pKa el número de moléculas ionizadas y no ionizadas es el mismo.
- Cuanto más alto sea el pH, mayor la ionización de fármacos ácidos y menor la de los básicos. Es decir, en medio basico los acidos se ionizan y no se absorben
- Cuanto más bajo sea el pH, menor la ionización de fármacos ácidos y mayor la de los básicos. Es decir, en medio acido, los acidos no se ionizan y se absorben
Para recordar formulas: Los acidos nos caen bien y son simetricos D/ND y las bases ND/D nos caen mal y son asimetricas. Un acido o base trata de neutralizar el medio, si están neutralizando el medio no pueden hacer dos cosas a la vez y disociarse tambien.
pH en distintos puntos del cuerpo:
- Estomago pH de 2 -> los acidos se absorben muy bien, las bases no
- Duodeno pH de 5,5 -> se aborben mas los acidos que las bases
- Tubulos renales pH de 6 -> se aborben mas los acidos que las bases
- Plasma de 7,4 -> bastante neutro, se disocian mas los acidos
- Colon pH de 8 -> se absorben mas las bases que los acidos
Hipótesis de reparto del pH: Cuando los valores de pH a ambos lados de una membrana lipídica son diferentes, los fármacos ácidos se encontrarán en concentraciones más altas en el lado más alcalino, en tanto que las básicas en el lado más ácido.
Esto es porque un acido en el medio acido no se disociara y por tanto atravesara la membrana y por tanto se encontrara en un medio mas alcalino en el que se disociara y ya no podra volver a atravesar la membrana quedandose atrapado y por tanto habra mas farmaco en la parte alcalina.
Factores que interfieren en la absorción de los farmacos:
- Diferencia de pH entre los lados de una membrana plasmatica habra distintas concentraciones a ambos lados y se quedaran atrapados en el compartimento con mayor diferencia con su pKa pasando a un estado estacionario.
- El atrapamiento ionico es el estado estacionario en el cual la concentración de los farmacos no ionizados es igual a ambos lados de la membrana plasmatica. El farmaco alcanzara mayor concentracion total en el compartimento en el que haya mayor fraccion ionizada
- El farmaco se acumula en el compartimento donde se encuentra su forma ionizada ya que no puede atravesar las membranas así.
- Aunque la disociacion y la ionización son conceptos diferentes, Una molecula disociada en este contexto significa lo mismo que una molecula no ionizada y viceversa.
Efecto de primer paso en la via oral: Inactivación a nivel hepático de algunos fármacos antes de que lleguen a circulación general.
Alteración de un fármaco por parte de las enzimas hepáticas antes de llegar a la circulación general. Los fármacos administrados en otras vías no pasan a la circulación porta para llegar al sistema circulatorio, por lo tanto, evitan el efecto de primer paso.
Factores que interfieren en la absorcion de los farmacos:
- Ionización completa del fármaco por sí mismo o por modificaciones del pH.
- Presencia de comida que disminuye el contacto del fármaco con la mucosa.
- Precipitación de una solución y formación de quelatos.
- Inactivación del fármaco por el pH, enzimas digestivas o metabolizadoras.
- Efecto de primer paso.
- Alteración del vaciamiento gástrico.
- Estimulación del peristaltismo intestinal.
- Interacciones que disminuyan la absorción.
Tema 5 - Farmacocinética - Distribución
Distribución: Es el proceso mediante el cual el fármaco se incorpora desde la circulación sanguínea hacia los distintos órganos y tejidos corporales, pasando a través de diversas membranas biológicas. Pueden estar libres o unidas a proteinas
Fracción libre
- Farmacologicamente activa
- Puede atravesar membranas (se difunde)
- Se metaboliza y produceunefecto
- Se excreta
- Fármaco disuelto en plasma
Fracción unida a proteinas:
- Farmacologicamente inactiva temporalmente
- Actúa como reservorio (aumenta la vida media del fármaco)
- Frecuente y variable según los fármacos
- Reversible
De las proteinas plasmaticas mas comunes:
- Albuminas se unen a los farmacos acidos como la penicilina o salicilatos
- Glicoproteinas acida alfa 1 se unen a las bases como propanol o lidocaina.
- Globulinas se unen a hormonas y vitaminas
- Tanto albuminas y globulinas
La fracción ligada a proteinas:
- Es inactiva ya que no puede alcanzar los tejidos
- Sirve como reservorio del farmaco ya que se libera con lentitud
- Las proteinas pueden saturarse, si la fracción libre aumenta demasiado se alcanzan niveles toxicos
- No cruzan las membranas por tanto ni llegan a tejidos ni cruzan la barrera hemato encefálica
- Se metaboliza y elimina con dificultad ya que no llegan facilmente a los organos que metabolizan y excretan
Los factores que alteran la union a proteinas plasmaticas es:
- Competencia por la misma proteina entre dos farmacos
- Disminucion de las proteinas (hipoproteinemia)
- Alteración de la calidad de las proteinas
Cuando tiene importancia clinica:
- Cuando afecta a la proteina especifica que fija al farmaco
- CUando afecta al lugar de fijacion proteina-farmaco
- Si el farmaco se une a mas del 80% de las proteinas plasmaticas
La mayoria de los farmacos atraviesan la pared capilar mediante difusion y filtración (poros)
La velocidad de salida depende de:
- Naturaleza del fármaco
- Tamaño
- Coeficiente de partición: Cuanto más alto es el cociente de partición, más lipo-soluble es el fármaco.
- Unión a proteínas plasmáticas
En los tejidos los farmacos están disueltos en el liquido intersticial e intracelular
Alcanzarán distintas concentraciones en unas u otras áreas del organismo dependiendo de:
- Flujo sanguíneo regional
- Especial afinidad por un tejido donde se acumula
- Masa de los distintos órganos en relación con el peso corporal
- pHdeloslíquidos corporales
- Liposolubilidad y grado de ionización
60% del peso corporal es agua
- 44% es intracelular
- 16% es extracelular - 4,5% Agua plasmatica, 9% agua intersticial y 2,5% agua transcelular
Volumen de distribución o Vd:
Es el volumen del líquido en el cual se encuentra distribuido o contiene el fármaco, este es aparente y no es real. Permite predecir la distribución de un fármaco en el organismo.
Cuanto mayor sea el Vd mas penetrara en los compartimentos, mayor concentración en tejido comprarada con la cantidad en sangre. Si el volumen de distribución es pequeño este estara confinada en la sangre ya bien porque el farmaco es hidrofilico, ionizado o unido a proteinas plasmaticas.
Modelos de distribución La farmacocinética considera al organismo dividido en compartimientos, acuosos o no, que se definen como sectores reales o virtuales del organismo al cual puede acceder o del cual puede salir un fármaco.
- El modelo monocompartimental considera que el farmaco se distribuye por todos los organos por igual
- El modelo bicompartimental considera que el farmaco se distribuye desigualmente. Se distribuye mas a los organos muy perfundidos como el higado, riñon, corazon, a estos se distribuye inmediatamente (compartimento central). En organos menos irrigados la distribución es mas lenta (Compartimento periferico).
Factores que modifican la distribución:
- Propiedades fisicoquímicas del fármaco: se distribuyen mejorlos medicamentos mas liposolubles, no ionizados y de bajo PM o peso molecular.
- Flujo sanguíneo del tejido: los fármacos alcanzan concentraciones más elevadas y con mayor rapidez en los órganos mejor perfundidos.
- Afinidad del fármaco por el tejido: por ejemplo, los digitálicos tienen afinidad por el tejido cardiaco y las tetraciclinas (La tetraciclina se acumula en dientes asi que puede dar color en los huesos si se usa donde el desarrollo )por el hueso.
- Contenido lipídico del tejido: existen fármacos muy liposolubles que se acumulan en la grasa quedando atrapados a ese nivel y son liberados con lentitud.
- Grado de unión a las proteínas plasmáticas: el grado de unión entre el fármaco y la proteína plasmática depende de:
- Naturaleza del fármaco
- Afinidad del fármaco para la proteína plasmática
- Disponibilidad de proteínas plasmáticas
Barreras corporales especiales: ofrecen dificultad al pasaje de algunos fármacos.
Barrera hematoencefálica (BHE):
- Solo es permeable asustancias liposolubles.
- Las sustancias muy ionizadas como las aminas cuaternarias o las penicilinas normalmente son incapaces de atravesarlas.
- Células epiteliales (vasos capilares SNC) muy unidas, no hay poros acuosos entre las células; impide difusión de sustancias polares de bajo PM.
- Células gliales (astrocitos) rodean los capilares del SNC.
Barrera hemato-placentarea
- Este órgano de intercambio materno-fetal consta de 3 estratos de tejidos fetales
- Epitelio trofoblástico
- Tejido conectivo coriónico
- Endotelio capilar
- La penetración es mínima para los fármacos con alto grado de disociación y baja liposolubilidad.
- Los amonios cuaternarios y las sustancias hidrosolubles de PM superior a 1.000 daltons no atraviesan la BP; sin embargo, la placenta es permeable a fracciones no ionizadas, a los no electrolitos liposolubles (éter, cloroformo), a las hormonas esteroideas, salicilatos, atropina, barbitúricos, antibióticos, alcaloides, etc.
- Los fármacos la atraviesan por difusión pasiva, facilitada y pinocitosis.
Las diferencias de distribución de un farmaco entre especies es debido a:
- Composición corporal (porcentaje de grasa y agua)
- Flujo sanguíneo y perfusión tisular.
- Niveles y tipos de proteínas plasmáticas.
- Permeabilidad de barreras biológicas (BHE, placenta).
- Afinidad tisular y actividad metabólica.
En la distribución no solo importa la dosis, sino a dónde llega el fármaco.
Tema 6 - Farmacocinética - Metabolismo
Metabolismo: La biotransformación es el conjunto de transformaciones químicas que sufre un fármaco dentro del organismo, catalizadas por enzimas, para hacerlo más hidrosoluble y fácil de eliminar. Busca la inactivación del farmaco (aunque no siempre es satisfactorio), lo hace volviendolo mas hidrosoluble e ionizado lo que permite una eliminación mas facil.
La biotransformación, en orden de importancia, se produce en:
- Higado
- Tubo digestivo (Jugo gastrico, enzimas digestivas y flora intestinal)
- Sangre (Gracias a hidrolasas y esterasas)
- Riñon
- Placenta
- SN central y periferico
Hay dos fases:
- Fase 1: Se añaden sustituyentes a la molécula o se liberan en ella grupos funcionales que aumentan la ionización e hidrosolubilidad. Es decir, se añade o quita algo
- Fase 2: usando la molecula resultante de la fase 1 se le aumenta el tamaño, inactivandola e incremando su solubilidad para su posterior eliminacion por la bilis y orina
Fase I: Reacciones de funcionalización:
Se produce mediante la adición de grupos funcionales: -OH, -COOH, -NH2, -SH2. Las reacciones microsomales se producen en el higado
Oxidación: La mas importante. Añade un grupo OH
- Realizada mayoritariamene por el reticulo endoplasmatico liso de los hepatocitos aunque tambien ocurre en otros tejidos en menor medida
- El sistema que lo permite es el Sistema de oxidación microsomal o sistema oxidasa de funció mixta que contiene el citocromo P450, necesita de un cofactor NADPH como donador de electrones y al oxigeno molecular como donante
- Hay dos tipos:
- Oxidación (sistema microsomal hepatico): Por Oxidación alifática, Hidroxilación aromática, N-desalquilación, O-desalquilación, S-desalquilación, N-oxidación y N-hidroxilación. Para recordar nombres, tiene la palabra oxidación, oxilación o desalquilación (S, O, N)
- Oxidación no microsomal, ocurre fuera del reticulo endoplamatico de los hepatocitos, en el citosol y mitcondrias de otras celulas. Se producen Oxidaciones de alcoholes y aldehídos y Desaminación oxidativa (MAO)
Reducción:
- No tan prevalente como la de oxidacion pero aun asi necesarias por si la de oxidación no vale, para poder seguir las reacciones de fase 2
- Las reacciones son: Azorreducción, Nitroreduccion,
- Se producen en el higado por el sistema enzimatico microsomal o a nivel intestinal por la flora bacteriana
- La oxidación y reducción alteran o crean nuevos grupos funcionales.
Hidrolisis:
- Se producen en sangre (un poco en higado) por la acción de enzimas
- Hidrosis de esteres (Esterasas) y amidas (amidasas)
- La hidrolisis rompe los enlaces esteres, amidas y peptidicos liberando nuevos grupos funcionales.
- Se encuentran en plasma, hematias y sistema microsomal hepatico ademas de en otros tejidos
Las reacciones de fase 1 o reacciones de funcionalización SIEMPRE Aumenta la polaridad de la molecula pero tambien pueden producir:
- Inactivación del farmaco
- Confierten un producto inactivo en activo (profarmaco)
- Confierten un producto activo en otro activo también (formación de metabolitos activos o toxicos)
Fase II: Reacciones de conjugación:
- Toman el resultado de la fase I
- Aumentan la polaridad aun mas mediante la union con moleculas endogenas lo que requiere energia
- Se producen con el farmaco o metabolito contienen hidroxilo (OH), carboxilo (COOH), Amino (NH2), Sulfhidrilo (SH) apropiado para combinarse con moleculas endogenas como el acido glucuronico, grupos sulfato, acetilo, metilo...
- Esto forma metabolitos polares, hidrosolubles e inactivos facilmente eliminables
- Hay 5 tipos de conjugación (glucoronico, sulfatica, acetilica, metilica y por aminoacidos)
Tipos de conjugación
- Conjugación con ácido glucurónico
- En la fracción soluble del higado donde se sintetiza acido urindindifosfato glucuronico o UDPGA a partir de glucosa
- El UDPGA es donador de acido glucuronico para conjugar farmacos/metabolitos
- Enzimas: UDP-Glucoroniltransferasa en microsomas hepaticos
- Farmacos/metabolitos han de tener uno de los 4 grupos funcionales
- Gran capacidad de glucuronidacion por multiples enzimas
- IMPORTANTE el gato es deficiente en glucoronidacion, riesgo de acumulación y toxicidad
- Conjugación con sulfato
- importante para los grupos fenolicos y grupos OH o hidroxilos alifaticos
- En fracción soluble del hepatocito
- Requiere la activación del grupo sulfato
- La enzima necesaria es la sulfotransferasa
- Es deficitaria en cerdo
- Conjugación por Acetilación
- Incorporación de radical acetilo (acetil-CoA) a grupos amino o carboxilo.
- Enzimas: acetiltransferasas.
- Reacción en la fracción soluble del hepatocito.
- Ejemplo: sulfamidas → metabolitos por acetilación.
- Deficitaria en perro
- Metilación
- Conjugación de un grupo metilo (de metionina).
- Se une a grupos -OH, -SH o -NH₂ del fármaco/metabolito.
- Enzimas: metiltransferasas.
- Localización: fracción soluble del hepatocito.
- Conjugación con aminoácidos
- Importante, la unica que se produce en mitocondrias
- Se unen a grupos COOH o carboxilos del farmaco o metabolito
- Se conjuga con aminoaciso como la glicina
Factores que modifican el metabolismo: Fisiologicos:
- Edad
- Especie
- Hormonales
- Genéticos
- Nutrición
Patologicos
- Hepatopatías
- Enfermedades hereditarias
Farmacologicos
- Inducción enzimática
- Inhibición enzimática
El paracetamol se bioactiva en primera fase por el citocromo P450 produciendo un metabolito toxico NAPQUI, la reaccion de glucoricacion lo desactiva.
Tema 7 - Farmacocinética - Excreción
Se denomina excreción de fármacos a la salida de estos y de sus metabolitos desde el sistema circulatorio al exterior del organismo.
La principal via de excreción es renal pero tambien hay via biliar, pulmonar y, en menor medida, pancreatica, digestiva, salivar y por el sudor/pelo
La excreción renal:
- 25% del gasto cardiaco, principal via de excreción
- Importante sobre todo en farmacos que se eliminan de forma inalterada (Sin pasar por la metabolización)
- Hay tres procesos, filtración glomerular (poros, transporte pasivo), secreción tubular (transporte PRINCIPALMENTE activo y pasivo), reabsorción tubular (farmacos liposolubles y transporte activo)
Filtración glomerular
- Transporte pasivo, la via mas importante de excreción
- Pasan 1200ml/min, de los cuales un 10% es filtrado, constituye la tasa de filtración glomerular
- El farmaco libre acompaña al filtrado y entra al espacio de bowman
- La solubilidad y pH no influyen, importante
- Factores que afectan a la filtración:
- Tamaño de la molecula y union a proteinas
- peso molecular (tiene que ser <20000 daltons, solo afecta a macromoleculas como la heparina
- La albumina no se filtra y los farmacos que se unen a ella tienen filtracion reducida, como en el caso de la warfarina
- Consecuencias:
- Los compuestos liposolubles y no ionizados se pueden reabsorber, los polares e hidrosolubles se filtran y dificilmente se reabsorben o secretan
- Se puede medir la filtración glomerular gracias a que la creatina e inulina no se unen a proteinas, se filtran pero no se reabsorben asi que sus valores de excrecion se usan como medidas de la tasa de filtración glomerular
Aclaramiento plamatico renal:
- Volumen de sangre deìradp de una sustancia por unidad de tiempo (ml/min). Se refiere a la capacidad de los riñones para eliminar una sustancia (inulina, creatinina) y nos sirven para medir la funcionalidad renal
Secreción tubular:
- Paso del farmaco desde el plasma desde los capilares peritubulares a la luz tubular (Transporte activo y pasivo)
- Transporte activo:
- Transportadores para ácidos débiles (aniones, carga negativa): furosemida, AINEs.
- Transportadores para bases débiles (cationes, carga positiva): morfina, antibióticos aminoglucósidos.
Reabsorción tubular:
- Difusión pasiva en el tubulo renal (reabsorción)
- 99% del agua se reabsorbe, solo un 1% del filtrado se queda
- Se reabsorben los farmacos liposolubles, no ionizados cuando el pH urinario es el adecuado
Exccreción biliar:
- El hígado excreta fármacos o metabolitos a través de la bilis hacia el intestino, desde donde pueden eliminarse en las heces o volver a la circulación.
- Sustancias eliminadas por vía biliar:
- Moléculas de elevado peso molecular (normalmente >300Da).
- Derivados conjugados que se forman por biotransformación hepática.
- Transporte activo transportadores específicos para ácidos, bases y sustancias neutras.
- Circulación enterohepática:
- Excreciónbiliar inicial
- Reabsorción del fármaco inalterado
- Reactivación de metabolitos conjugados (glucuronidasas).
- Importante, Los fármacos que sufren ciclo enterohepático retrasan su eliminación y, por tanto, aumentan la duración de su efecto terapéutico/tóxico.
Otras vias de excreción:
- Pulmón = Gases y vapores volátiles (anestésicos inhalatorios: isoflurano, sevoflurano)
- Leche = pH menor que plasma, se produce el atrapamiento ionico de las sustancias basicas. Importante en producción animal
- Heces = Vía biliar/entérica. Fármacos administrados por vía oral que no se absorben. Acceso desde la sangre al intestino.
- Secreciones glandulares: Poco importante. Monitorización de los niveles de fármacos en saliva.
- Piel y faneras: Eliminación de metales pesados como arsénico, mercurio. Diagnóstico de intoxicaciones y medicina forense.
Tema 8 - Farmacocinética Cuantitativa
La farmacocinética cuantitativa nos permite, mediante modelos matemáticos sencillos, conocer los siguientes parámetros farmacocinéticos:
- Velocidad de absorción
- Cantidad de fármaco absorbido
- Modo de distribución del fármaco
- Velocidad de eliminación
Esto nos permite establecer un regimen posológico, encontrar la dosis optima, intervalo de administración, duración del tratamiento
Curvas de nivel plasmatico: Representan la evolución de la concentración de fármaco en plasma a lo largo del tiempo desde su administración hasta su excreción. Cuando un fármaco es administrado vía endovenosa no sufre el proceso de absorción. Nos dice:
-
Cuándo empieza el efecto.
-
Cuánto tiempo se mantiene en niveles eficaces.
-
Cuándo desaparece el fármaco del organismo
-
Cmax (Concentración máxima): Es la concentración plasmática más alta que alcanza el fármaco tras su administración. 👉 Indica la intensidad máxima del efecto.
-
Tmax (Tiempo hasta la concentración máxima): Es el tiempo que tarda en alcanzarse la Cmax. 👉 Refleja la velocidad de absorción del fármaco.
-
CME (Concentración mínima eficaz): Es la concentración plasmática mínima necesaria para que el fármaco produzca un efecto terapéutico. 👉 Por debajo de este valor, el fármaco no tiene acción clínica.
-
CMT (Concentración mínima tóxica): Es la concentración plasmática mínima a partir de la cual aparecen efectos adversos o tóxicos. 👉 Por encima de este valor, el fármaco deja de ser seguro.
-
Rango terapéutico: Es el intervalo entre la CME y la CMT. 👉 Dentro de este rango, el fármaco es eficaz y seguro.
-
Periodo de latencia: Es el tiempo que transcurre desde la administración hasta que aparece el efecto clínico. 👉 Depende de la absorción y distribución inicial.
-
Tiempo que pasa desde la administración hasta el efecto: Es básicamente el inicio de acción, que coincide con el momento en que la concentración plasmática supera la CME.
La farmacocinética considera al organismo dividido en compartimientos, acuosos o no, que se definen como sectores reales o virtuales del organismo al cual puede acceder o del cual puede salir un fármaco.
Modelo monocompartimental:
- El mas sencillo, considera solo un compartimento homogeneo.
- El fármaco, tras su administración, se distribuye rápida y uniformemente.
- La concentración plasmática desciende de manera exponencial según se va eliminando.
- Es útil para la mayoría de los fármacos de uso común, y como punto de partida para entender modelos más complejos.
- En graficas intravenosas son siempre descendentesy plenamente representativas de los procesos de eliminación
Conceptos importantes de los modelos farmacocineticos:
-
Constante de eliminación (Ke)
- Indica la fracción del fármaco que se elimina por unidad de tiempo.
- Se mide en tiempo⁻¹ (ej. h⁻¹) (tiempo recíproco).
- Relaciona la concentración remanente en un tiempo dado con la que existe en la unidad de tiempo inmediatamente anterior.
- En una grafica seria la pendiente de la recta
- Ke= ln C0 (logaritmo neperiano de la concentración plasmatica inicial) - LnCp (Logaritmo neperiano de la concentración en un tiempo determinado) / t, tiempo transcurrido entre C0 y Cp)
-
Tiempo de vida media (T1/2)
- Tiempo necesario para que la concentración plasmática de fármaco se reduzca a la mitad.
- Se expresa en unidades de tiempo real no en tiempo recíproco como la constante de eliminación.
- Nos sirve para establecer pausas posologicas
- T1/2 = ln2 (logaritmo neperiano de 2, es decir 0,693) / ke o constante de eliminación
-
Volumen de distribución (Vd)
Factores que modifican la vida media
- Dosis y coadministración de fármacos: Algunos fármacos inducen o inhiben enzimas hepáticas.
- pH urinario
- Ácidos débiles → se eliminan más si la orina es básica (se ionizan más).
- Bases débiles → se eliminan más si la orina es ácida (se ionizan más).
- Especie, raza y edad
- Gatos (aspirina), Collies (mutación MDR1), jóvenes o geriátricos.
- Patologías
- Insuficiencia hepática, insuficiencia renal vida media más larga.
- Deshidratación (aclaramiento plasmático más lento) vida media más larga.
Volumen de distribución (Vd)
- Es una constante de proporcionalidad que relaciona la cantidad de fármaco existente en el organismo en un instante dado con su concentración plasmática en el mismo instante.
- Unidades: litros ó litros/kg p.v
- Vd alto: indica que el fármaco se distribuye ampliamente en tejidos (ej. lipofílicos, que se acumulan en grasa o músculo) o que la distribución no es instantanea (modelo bicompartimental)
- Vd bajo: significa que el fármaco se queda principalmente en el plasma (ej. fármacos muy unidos a proteínas plasmáticas).
- Si el Vd es 4,5%= se queda en la sangre, si es 13%-16 llega al intersticial 1 si es 60% se distribuye en todo el cuerpo
𝑉𝑑 = 𝐷/𝐶0
Aclaramiento plasmatico (Cl)
- Es el volumen de plasma depurado de fármaco por unidad de tiempo por el conjunto de los órganos eliminadores del organismo.
- Unidades: volumen/tiempo
- Cl = Vd * Ke siendo Ke la constante de eliminación o tambien vale Cl = Vd * 0,693 (ln 2) / t 1/2
- Relaciona la capacidad de eliminación con el volumen donde está distribuido el fármaco.
- Es una fórmula teórica, matemática, derivada del modelo monocompartimental.
- Es la deducción matemática a partir de cómo cae la concentración plasmática con el tiempo (curvas plasmáticas).
- Cl =Vd * Cu o concentración en orina / Cp o concentración plasmatica
- Mide directamente qué volumen de plasma queda “limpio” de fármaco por minuto en función de lo que aparece en orina.
- Es una fórmula práctica y experimental, útil en estudios de balance.
- Mide experimentalmente lo que sale por orina en relación con lo que había en plasma.
La curva en vias extravasculares es ascendente hasta alcanzar Cmáx siguiendo la constante de absorción o Ka (cuando la velocidad de absorción = velocidad de eliminación) y luego descendente.
Velocidad de absorción Ka
- La concentración de fármaco que se absorbe por unidad de tiempo va disminuyendo exponencialmente a medida que disminuye la concentración del fármaco en el compartimento del que desaparece.
- Se mide en tiempo⁻¹ (ej. h⁻¹) (tiempo recíproco).
- Ejemplo: ka = 0,05 h-1 indica que en 1 hora se absorberá el 5 % de las moléculas del fármaco en disolución.
Semivida de absorción T 1/2 a
- Tiempo que tarda en reducirse a la mitad el número de moléculas de fármaco que quedan por absorberse. T1/2 a = 0,693/ka
- Para medirlo hay metodos directos o indirectos:
- Metodo directo: Metodo de los residuales: restar del nivel plasmático teórico que existiría de no haber mediado absorción, el valor obtenido experimentalmente en el mismo instante.
- Esta diferencia, representa la fracción que no ha pasado al plasma y permanece en el lugar de absorción. Se realiza extrapolando la fracción recta de la representación semilogaritmica de las curvas de nivel plasmatico, calculando las diferencias entre concentraciones extrapoladas y las experimentales, se representan las diferencias en la grafica y se mide la diferencia. La pendiente de la curva será ka.
Biodisponibilidad (F o Fraction, Absorbable Fraction ):
- Es la cantidad de fármaco absorbida en relación a la cantidad de fármaco administrada. En intravenosa es el 100%
- Se calcula el área bajo la curva (ABC) de Cp del fármaco: ABC = C0/ke
- Se expresa en % en relación a la vía intravascular: biodisponibilidad del 100 %.
- Para el mismo principio activo: comparamos el ABC por diferentes vías de administración y formas farmacéuticas con ABCiv = 100 %, y nos permite determinar la dosis adecuada.
- Biodisponibilidad en % = Area bajo la curva extravascular / Area bajo la curva intravenosa x 100
Bioequivalencia
- El parámetro más restrictivo.
- Requisito para especialidades farmacéuticas genéricas (EFG).
- Un fármaco genérico, además de la misma formulación y principio activo que otro fármaco ya comercializado ha de ser bioequivalente:
- Se acepta un maximo de un 20% de diferencia entre area bajo la curva, C max y T max
Influencia en la vía de administración
- El tramo descendente exponencial tiene la misma forma en todas las curvas (ke y t1/2 mismo valor para todas).
- El tramo exponencial se desplaza a la derecha a medida que la aparición del fármaco en sangre se hace más lenta (el fármaco permanece más en el plasma cuanto más lenta es su absorción).
- El valor de la intersección de la recta de la fase terminal extrapolada en el eje de ordenadas es mayor cuanto más lenta es la absorción (el antilog de C0 aumenta a medida que Ka disminuye). Las rectas de pendiente Ke son paralelas.
- El valor máximo de Cmax es más bajo y la curva es más roma cuanto más lenta es la absorción.
- La curva IV corta a todas en sus máximos. Absorción completa y dosis iguales.
- El valor de ABC de niveles plasmáticos es constante, sea cual fuere la administración utilizada si la biodisponibilidad es equivalente.
Régimen de dosificación racional
- Es la pauta de administración de medicamento mediante la cual se alcanza un nivel terapeútico eficaz en la biofase (compartimento funcional donde el fármaco actúa), se mantiene éste durante todo el tratamiento y se previene la acumulación del medicamento en el organismo.
- Los parametros que lo rigen son:
- C min: Concentración plasmática mínima eficaz o CME
- C max: Concentración plasmática, inferior a la concentración plasmática tóxica, ha de estar por debajo de CMT
- D: Dosis de mantenimiento sensiblemente igual a la dosis mínima eficaz, repetida a intervalos fijos
- D*: Dosis de choque inicial: es el doble de la de mantenimiento, pero solo se utiliza para la primera vez. Se usa para alcanzar mas rapidamente un nivel estable de concentración minima eficaz.
- T: Intervalo de dosificación o tiempo transcurrido entre dos administraciones.
Dosis de mantenimiento vía oral
- D: dosis
- T: Intervalo de dosificación o tiempo transcurrido entre dos administraciones.
- D/𝑻 : tasa de dosis por intervalo de administración (dosis/horas; dosis/días)
- Cmss o Concentracion Media Steady State: Concentración del fármaco media deseada en equilibrio estacionario.
- F: biodisponibilidad del fármaco por vía oral.
- Cl: aclaramiento del fármaco
- Con esta fórmula establecemos la dosis/tiempo
Para calcular una pauta de dosificación
D/T = Cobjetivo o Cmss x Cl o aclareamiento / F o Biodisponibilidad
Cuando T = t1/2 -> Cmax/Cmin ≈ 2
Tema 9 - Farmacodinámia
Lo que el farmaco le hace al organismo:
Estudia los acontecimientos derivados de la interacción entre el fármaco y su receptor u otro lugar primario de acción. Estudia los efectos bioquímicos y fisiológicos de los medicamentos, su mecanismo de acción y la correlación entre las acciones y efectos de los medicamentos y la estructura química.
La acción farmacolofica modifica función, nunca la crea. Puede:
- Estimular
- Inhibir
- Reemplazar (insulina)
- O ser un Antimicrobiano
Los mecanismos de acción Pueden ser por union a receptores
- Unión a receptores: Activa procesos bioquimicos o bloquea su funcionamiento.
Mecanismos que no involucran a receptores:
- Tienen efecto sobre mecanismos fisico-quimicos generales:
- Agentes quelantes como el EDTA
- Surfactantes (modifican caracteristicas de las membranas o liquidos) como el docusato sodico
- Adsorbentes como el carbon activado
- Antiacidos como el hidroxido de aluminio
Interacción farmaco - receptor
- Especifidad: Capacidad para actuar únicamente o predominantemente sobre un receptor concreto.
- Reversibilidad: La unión fármaco-receptor puede no ser permanente.
- Saturabilidad: El número de receptores disponibles es limitado.
- Efecto intracelular: La unión del fármaco al receptor desencadena una cascada de señales bioquímicas dentro de la célula, que conllevan el efecto terapéutico.
Importancia de los receptores
- Determinan la relación entre la dosis o concentración de un fármaco y su efecto.
- La selectividad de acción farmacológica depende de ellos y de su interacción con los fármacos a los que se liga.
- Modulan las acciones de agonistas y antagonistas farmacológicos.
Clasificación de los receptores:
- 7 grandes familias con distintos tiempos de respuesta.
- Hay inotropicos (rápidos y ligados a canales iónicos.) y metabotropicos (más lentos y ligados a cascadas de señalización intracelular.)
Receptores acoplados a proteínas G (GPCRs)
- Los mas frecuentes en mamiferos y hay 800 tipos. El 40% de los farmacos actuan en base a los GPCR
- Son receptores transmembrana que activan segundos mensajeros (ej.: AMPc, IP3).
- Receptores de histamina, dopamina, opioides, muscarínicos de Ach, adrenérgicos…
- Ejemplos:
- Clembuterol activa receptores β2-adrenérgicos en el músculo liso bronquial de caballos produciendo broncodilatación.
- Morfina estimula receptores opioides μ en el SNC en perros y gatos induciendo analgesia.
Canales iónicos transmembrana
- Proteínas transmembrana que forman un poro selectivo y cuando el ligando se une permiten el flujo de iones en la célula y organelas a favor de gradiente.
- Tres grupos:
- Cambios de voltaje. (Canales de Na+, canales de K+, canales de Ca+2).
- Ligandos o segundos mensajeros. Neurotransmisores. (Canales de GABA, nicotínicos, de serotonina (5-HT3), ionotrópicos de glutamato (NMDA, AMPA…)).
- Otros (acuaporinas, conexinas, canales de Cl-). No abren directamente el canal sino que son creados por otros mecanismos y modulan la actividad del canal desde dentro de la celula ??? IMPORTANTE, REVISAR
- Ejemplos: Lidocaína, benzodiacepinas, ketamina.
Receptores catalíticos
- Receptores con actividad enzimática.
- Tirosina-cinasa.
- “Receptores transmembrana con dominios citosólicos con actividad enzimática” Tienen una parte extracelular que reconoce al ligando y una intracelular con función enzimática.
- Fosforilación (cinasa) o una desfosforilación (fosfatasa).
- Ejemplo: receptor de insulina en el tratamiento de la diabetes mellitus en perros.
Receptores nucleares
- Localizados en núcleo.
- Regulan transcripción génica.
- Promueven o reprimen la transcripción de sus genes diana en respuesta a la unión de sus ligandos.
- Dos grupos:
- Receptores de esteroides (estradiol, cortisol, corticosterona, progesterona…) y
- Receptores de hormonas no esteroideas (tiroideas, vitamina D…).
- Ejemplo: los glucocorticoides (p. ej.: prednisolona) se unen a receptores nucleares produciendo efectos antiinflamatorios o inmunosupresores.
Enzimas
- Enzimas extracelulares como diana farmacológica.
- Ejemplos:
- IECAs (Inhibidores de la Enzima Convertidora de Angiotensina)
- Bloquean la enzima que convierte angiotensina I en angiotensina II (una sustancia vasoconstrictora) vasodilatación, reducción de presión arterial.
- Ejemplo: tratamiento de insuficiencia cardíaca en perros.
- Fármacos anticolinesterasa
- Inhiben la acetilcolinesterasa, enzima que degrada la acetilcolina aumenta la concentración de acetilcolina en la sinapsis se prolonga su efecto.
- Ejemplo: neostigmina en casos de íleo paralítico en caballos o en el diagnóstico/tratamiento de miastenia gravis.
- Organofosforados
Cinasas intracelulares
- Fosforilación para modificar la actividad de proteínas, activándolas o desactivándolas.
- Papel central en la función de GPCRs.
- Participan en cascadas de señalización (transducción de señales).
- Ejemplo: toceranib (Palladia®) para el tratamiento de mastocitomas caninos.
Transportadores
- Proteínas que mueven sustancias a través de membranas celulares.
- Ayudan a los compuestos polares o cargados a atravesar membranas biológicas, que son hidrófobas.
- Dos grupos:
- SLC (transportadores de solutos)
- ATPasas
- Ejemplo: la furosemida bloquea el transportador Na⁺-K⁺2Cl⁻ (NKCC2) que se encuentra en la rama ascendente gruesa del asa de Henle, impidiendo que esos iones se reabsorban efecto diurético.
Otros: Proteínas de adhesión, Receptores sigma, Chaperonas(asistir en el plegamiento correcto de otras proteínas), Claudinas, Proteínas de choque térmico, Cinesinas, Inmunoglobulinas, Factores de intercambio guaninas, Antígenos de linfocitos, Tubulinas, Factores ribosómicos.
Conceptos de la unión farmaco - receptor:
- Afinidad: Mide el grado de union de un farmaco a su receptor
- Eficacia (e): Capacidad de un farmaco para producir una respuesta una vez unido al receptor (no la tienen los antagonistas ya que no producen respuesta)
- Actividad intrinseca (a o alfa): La respuesta que produce un agonista con respecto a la respuesta maxima que produce un agonista completa
Los farmacos que se unen a un receptor pueden ser:
- Agonistas
- Puro/total
- Parcial
- Inverso
- Antagonistas
- Competitivo
- No competitivo
Agonistas: tienen afinidad y actividad intrinseca
- Agonista total: se une al receptor y lo activa completamente, imitando al ligando endógeno. Ej.: epinefrina.
- Agonista parcial: se une al receptor, lo activa parcialmente, y nunca logra un efecto máximo, aunque todos los receptores estén ocupados. Ej.: buprenorfina.
- Agonista inverso: se une al receptor y reduce su actividad basal.
Antagonistas: solo tienen afinidad, no tienen eficacia ni actividad intrinseca
- Antagonista competitivo: bloquea el efecto del agonista compitiendo por el mismo sitio de fijación. Ej.: naloxona para revertir sobredosis de opioides.
- Reversible: puede ser desplazado del receptor por dosis crecientes del agonista.
- Irreversible: no puede ser desplazado del receptor por dosis crecientes del agonista.
- Antagonista no competitivo: bloquea el efecto del agonista uniéndose al receptor en un sitio distinto al sitio de fijación del agonista. Ej.: ketamina.
- Reversible: se disocia fácilmente del receptor al suspender su administración al paciente.
- Irreversible: se fijan permanentemente o modifica covalente el receptor el cual queda permanentemente inutilizado y tiene que ser reemplazado por uno nuevo.
Diferencia entre un antagonista y agonista inverso: El antagonista es “neutral”: bloquea pero no genera respuesta. El agonista inverso sí genera respuesta, pero en dirección contraria a la del agonista.
La curva dosis respuesta
- Representa la relación entre la dosis administrada de un fármaco y la respuesta farmacológica.
- Tiene forma sigmoidea
- Sus caracteristicas potencia, actividad intrinseca y seguridad
- La dosis umbral es aquella en la que se empieza a ver respuesta
- Emax es la respuesta maxima que puede producir
- Actividad intrinseca: determinada por la altura de la Emax, nos indica la eficacia, la altura lo determina, cuanto mas arriba mas eficaz
- Potencia: cuanta mas potencia mas rapidamente alcanzara un efecto, es decir, menos dosis se necesitará. Cuanto mas a la izquierda mas potente
- Indice terapeutico: relación entre la toxicidad (dosis letal) y la eficacia (dosis eficaz) Se calcula IT con IT = DL50 / DE50 NO DOSIS TOXICA
- DE50 (Dosis Efectiva 50): la dosis que produce el 50% del efecto máximo. Es el punto que nos determina la potencia de un farmaco
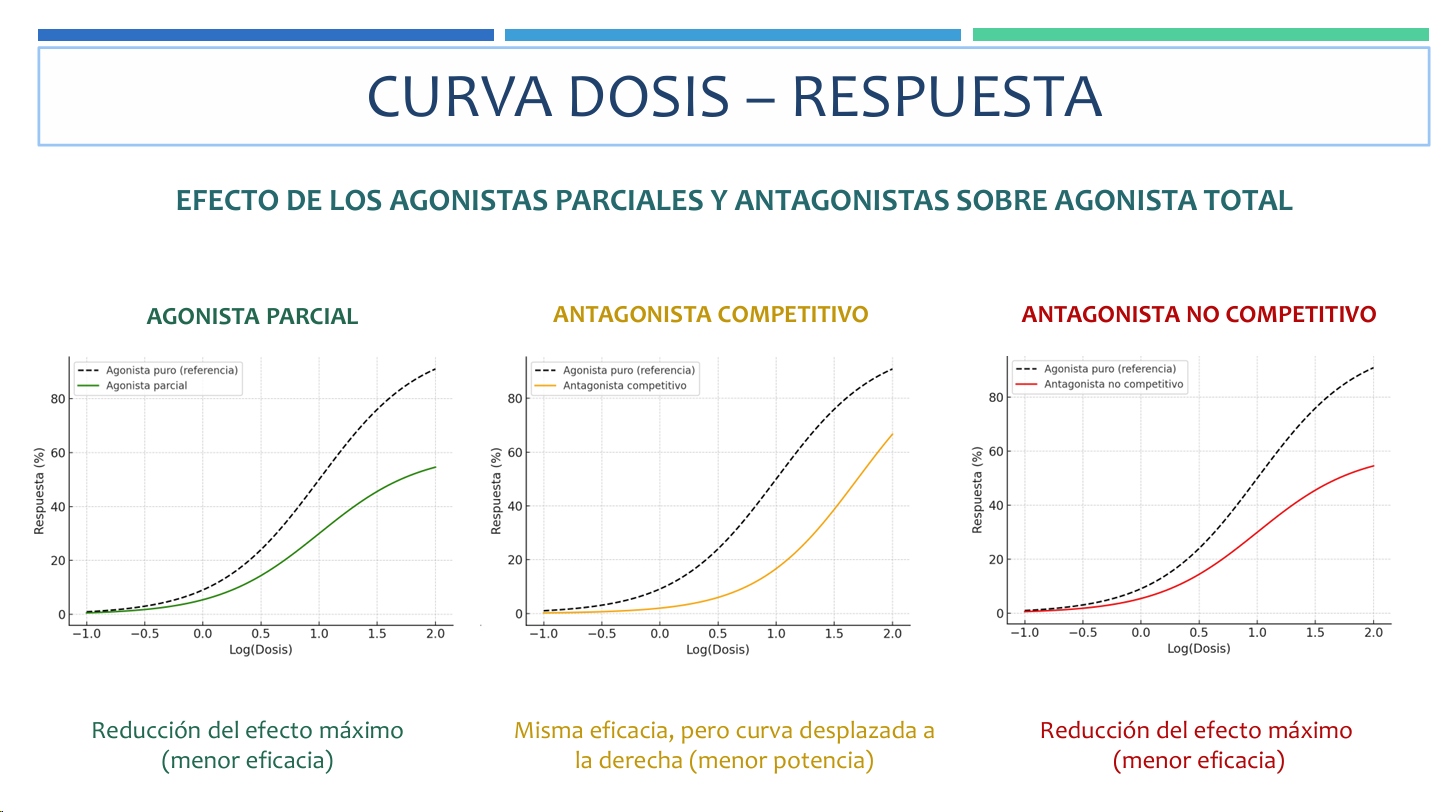
IMPORTANTE, un antagonista competitivo solo desplaza la cuva a la derecha, es decir, reduce la potencia mientras que un antagonista no competitivo reduce el efecto maximo, es decir, reduce la eficacia reduciendo la altura de la curva
Interacciones farmacodinamicas: Dos o más fármacos actúan sobre un mismo sistema fisiológico o ruta bioquímica, afectando el efecto terapéutico de uno u otro, ya sea potenciándolo o reduciéndolo.
Sinergismo: Los efectos que se obtienen son iguales o superiores a la suma de sus efectos por separado IGUAL O SUPERIOR
- Sinergismo de adición: el efecto de dos fármacos administrados conjuntamente es igual a la suma de los efectos individuales.
- Sinergismo de potenciación: el efecto de dos fármacos administrados conjuntamente es superior a la suma de los efectos individuales.
Antagonismo: Disminución o anulación del efecto de un fármaco por la acción de otro.
- Antagonismo competitivo: ambos compiten por el mismo receptor.
- Antagonismo fisiológico: los fármacos actúan sobre diferentes receptores pero con efectos opuestos.
- Antagonismo químico: un fármaco neutraliza químicamente al otro.
Tipo de efecto indeseado:
- Sobredosis Exceso de dosis Paracetamol en gatos
- Efecto colateral - Esperado, pero no deseado - Hipotensión con acepromazina
- Efecto secundario - Consecuencia del mecanismo - Estreñimiento con opioides
- Reacción alérgica - Inmunológico - Urticaria con penicilina
- Tolerancia - Respuesta disminuida - Fenobarbital en perros
- Taquifilaxia - Pérdida rápida de efecto - Efedrina
- Idiosincrasia - Respuesta genética anómala - Ivermectina en Collies